Fig. 10.1
Excitation-contraction coupling in cardiac muscle. The initial event in the cardiac muscle contraction is membrane depolarization, which occurs with ion entry through connexon channels from a neighboring cardiomyocyte, followed by opening of voltage-gated Na+ channels and Na+ entry. The resultant rapid depolarization of the membrane inactivates Na+ channels and opens both K+ channels and Ca2+ channels. Entry of Ca2+ into the cell triggers the release of Ca2+ from the sarcoplasmic reticulum through the ryanodine channel. Ca2+ then binds to the troponin complex and activates the contractile apparatus. Cellular relaxation occurs on removal of Ca2+ from the cytosol by the Ca2+-uptake pumps of the sarcoplasmic reticulum and by Na+/Ca2+ exchange with the extracellular fluid
10.2 Cardiac Hypertrophy
Hypertrophic growth of the heart is considered an adaptive response to pressure or volume overload, mutations of sarcomeric (or other) proteins, or loss of contractile mass from myocardial infarction. However, over time, the heart loses its ability to keep up with the chronic metabolic demands of increased mechanical workload, and dilation and heart failure (HF) ensue [5]. For this reason, myocardial hypertrophy is considered to be the interface between a normal and failing heart.
There are two types of hypertrophy: concentric and eccentric. Chronic left ventricular (LV) pressure overload (afterload) results in a concentric type of hypertrophy, where new sarcomeres are added in parallel to existing sarcomeres, and consequently, the wall thickness becomes increased [6]. Concentric Hypertrophy typically occurs in cases of aortic stenosis and hypertension and is believed to function by diminishing wall stress and oxygen consumption [7]. It is characterized by normal peak systolic and end diastolic wall stresses, with little or no change in chamber volume, but with increased h (wall thickness)/R (radius) ratio [8]. In contrast, eccentric hypertrophy, which increases the number of sarcomeric units in series, occurs in response to chronic LV volume overload (preload) and is typically characterized by an enlarged LV chamber [6]. Eccentric hypertrophy is typically observed in patients with aortic or mitral valve insufficiency. It induces an increased end diastolic wall stress, normalizes h/R ratio, and in general, presents with normal peak systolic wall stress, thereby increasing the shortening capacity of the myocyte and preserving LV function [8]. However, despite this, eccentric hypertrophy with elongation of myocytes can be a hallmark of end-stage pressure overload hypertrophy as well. Therefore, it is important to distinguish between adaptive elongation of the cardiomyocyte and elongation associated with failure [9].
10.2.1 Hypertrophic Cardiomyopathy
The principal anatomical feature of pathological growth of the heart is referred to as hypertrophic cardiomyopathy (HCM), an abnormal, asymmetric LV thickening of the heart [4]. Specifically, pathological hypertrophy is defined as an increase in the overall size of cardiac cells, with increased myocardial fibrosis that is distributed throughout the interstitium and in discrete foci [10]. Data suggest a remarkable and characteristic cardiac histopathology in HCM; hearts have misaligned myocytes with enlarged nuclei and expanded interstitial matrix, combined with both increased perivascular and interstitial fibrosis [11]. Interestingly, HCM usually affects the interventricular septum more than the LV–free wall [11].
Cardiac hypertrophy can also occur as a consequence of genetic alterations. Specific, autosomal dominant sarcomeric mutations can cause HCM, with over 400 mutations in at least 13 different sarcomeric proteins identified thus far [12]. Mutations in β-myosin heavy chain myosin (βMHC) were the first to be identified in HCM and cause ~30 % of all cases [13]. Mutations in α-cardiac actin, tropomyosin, and troponin have also been associated with cardiomyopathy [11].
Cardiac hypertrophy can also be caused by ischemic disease, hypertension, HF, and valvular disease [10]. When an increased work load is chronically imposed on the myocardium, such as by external stimuli and/or pathological stress such as hypertension, heart muscle injury (myocardial infarction), or neurohumoral stimulation, there is increased adenosine triphosphate (ATP) utilization by cardiac cells, increasing energy consumption of the heart and reducing inotropic reserves [6]. However, over time, this increased signaling induces pathological remodeling of the heart, causing the heart to become abnormally thickened or hypertrophic [12]. When the heart can no longer keep up with this excess demand, it decompensates, transitioning to dilation and, ultimately causing end-stage HF [12], a serious life-threatening condition and one of the most common causes of death worldwide [4].
10.2.2 Pathological Signaling Pathways
Several types of pathological conditions induce cardiac hypertrophy by activating a variety of membrane receptors to engage downstream intracellular signaling pathways, including the G-protein coupled receptor (GPCR), receptor tyrosine kinase (RTK), cytokine, and calcium-modulated pathways [14]. Acutely, the purpose for the activation of these pathways is to enhance excitation-contraction coupling, to allow for the heart to compensate for the excess demand [2, 3] (Fig. 10.1). Chronically, however, constitutive activation of these signaling pathways leads to sustained hypertrophic responses that induce maladaptive remodeling of the heart.
Interestingly, there is significant cross-talk between the proteins within these various signaling cascades, allowing for specific and directed responses to downstream effectors [15]. For example, stimulation of the mitogen activated protein kinase (MAPK) cascade by each of the three family members, the extracellular regulated kinases (ERK1/2, ERK5), p38 and the c-Jun N-terminal kinase (JNK), culminates in the activation of nuclear transcription factors that drive hypertrophic responses within the cell [16]. Increases in intracellular Ca2+, which leads to the activation of calmodulin-dependent protein kinase II (CaMKII) and/or calcineurin, induces activation of the nuclear factor for activation of T cells (NFAT), enhancing cardiac hypertrophy [17]. In addition, increased CaMKII activity modulates hypertrophic gene transcription by controlling nuclear shuttling of class II histone deacetylases (HDACs) [18]. Therefore, it is the concomitant increase in multiple signaling cascades that ultimately leads to the panoply of responses that drive enhanced (and chronic) hypertrophic gene transcription [19].
10.2.2.1 G-Protein Coupled Receptor Signaling
GPCRs are the largest class of cell surface receptors, with nearly 800 family members identified to date [20]. The GPCR consists of a cytosolic region, a ligand-binding extracellular region and seven transmembrane domains. Intracellularly, GPCRs are associated with heterotrimeric GTP-binding proteins (G-proteins) consisting of multiple isoforms of distinct Gα, β and γ subunits [21]. There are four sub-classes of G-proteins: Gαs and Gαi, which either stimulate or inhibit adenylyl cyclase (AC), respectively; Gαq/11, which activates phospholipase C (PLC); and Gα12/13, which activates the Rho family of G proteins [22]. Because the signal transducing properties of the various possible βγ combinations do not appear to radically differ from one another, these classes are defined principally according to the isoform of their α-subunit; however, βγ dimers are important in that they are involved in enhancing the complexity of possible interactions between G proteins and their targets [23].
In the absence of signal, the GPCR-associated heterotrimeric G protein is bound to guanosine diphosphate (GDP) and is inactive. However, upon binding of the agonist to the GPCR, a conformational change takes place which enables the receptor to act as its own guanine nucleotide exchange factor (GEF), activating the associated G-protein by exchanging its bound GDP for guanosine triphosphate (GTP) [24]. The G-protein’s α subunit then, together with the bound GTP, dissociates from the βγ subunit to further affect intracellular signaling proteins or target functional proteins directly dependent on the α subunit type [22]. GPCR activation drives downstream signaling cascades controlled by ACs, phospholipases and ion channels, as well as by various RTK cascades (ERK, JNK, p38, ERK5) and the phosphatidylinositol 3′ kinase (PI3K/Akt) pathway. In fact, GPCR signaling controls most important and vital molecular processes, such as cell proliferation, differentiation, survival, migration and other functions; however, chronic activation can also lead to the development of cardiovascular disease and HF [25].
10.2.2.2 Gαs and Gαi Signaling
β-1 adrenergic receptors (B1AR), β-2 adrenergic receptors (B2AR), dopamine-1 receptors, histamine-2 receptors, and vasopressin-2 receptors are examples of Gαs protein-coupled receptors [26]. Upon agonist binding, dissociated Gαs (“s” for stimulatory) leads to stimulation of AC and increased generation of cyclic adenosine 3′ 5′ monophosphate (cAMP), a key second messenger that mediates excitation-contraction coupling of the heart [27, 28]. The cAMP subsequently mediates activation of the serine/threonine-specific protein kinase A (PKA), which is ultimately required to modulate cardiac contractility via intracellular Ca2+ movements [26] (Fig. 10.2).
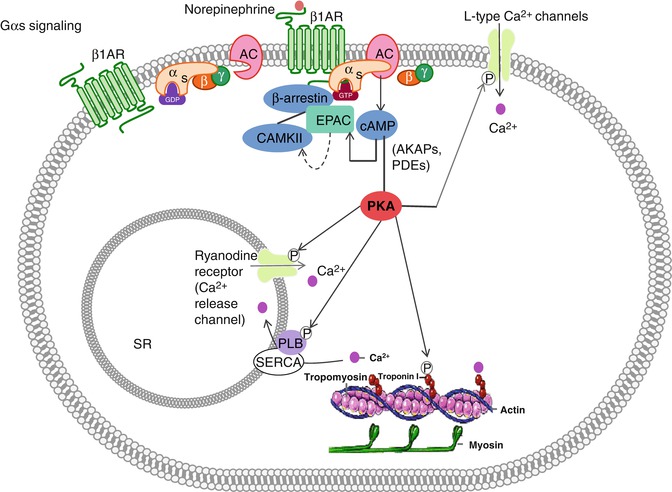
Fig. 10.2
Gαs signaling. When no signal molecule is present, the G protein binds to the GDP and is inactive. However, upon binding of a signal protein to the ligand-binding site, a conformational change of the receptor takes place. This enables the receptor to interact with the α subunit of the associated G protein, which then exchanges its bound GDP to GTP. This activates the G protein and causes βγ subunit to dissociate, thus enabling it to relay the signal by regulating the activity of additional intracellular signaling proteins. When norepinephrine (NE) binds to the β1AR, the G protein becomes activated and adenylate cyclase becomes stimulated. Cyclic AMP increases and phosphorylation events inside the cell take place, leading thus to increased cellular contraction. A β arrestin dependent scaffold including CAMKII and EPAC can be recruited to β1AR on stimulation, allowing cAMP-EPAC-mediated activation of CAMKII and regulation of contractility
Once activated, the catalytic subunit of PKA phosphorylates a range of substrates within the cardiomyocyte, including cardiac troponin I (cTnI), the ryanodine receptor (RyR), the LTCCs, and phospholamban (PLB), all of which are required to enhance cardiomyocyte contractility through modulation of Ca2+ transients released from the SR [26] (Fig. 10.2). In addition, PKA activity is regulated by A-kinase anchoring proteins (AKAPs) and phosphodiesterases (PDEs) to similarly modulate cardiomyocyte contractility and function [29].
AC activity is inhibited by stimulation of the Gαi-coupled muscarinic acetylcholine receptor-2 (mAchR2) as well as by stimulation of the sphingosine-1 phosphate receptor-1 (S1PR). In addition, Gβγ subunits, when released from the Gα subunits, function to open muscarinic K+ channels (Kach) and promote membrane hyperpolarization of the cardiomyocyte. Reduction of the action potential duration follows, and chronotropy and inotropy are decreased [30] (Fig. 10.3).
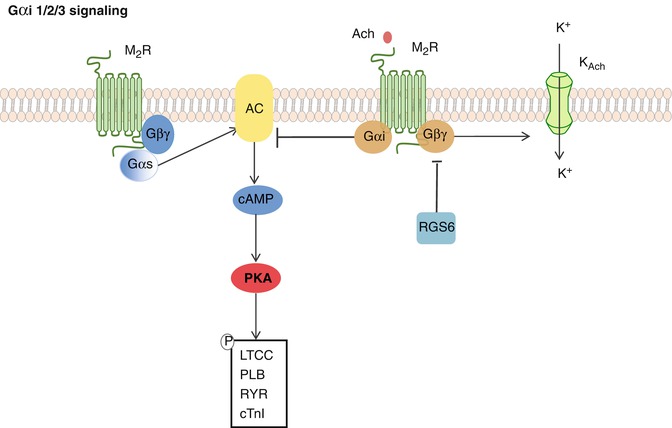
Fig. 10.3
Gαi 1/2/3 signaling. The muscarinic acetylcholine receptor 2 is Gαi coupled, and upon its stimulation, AC activity is decreased. Moreover, the Gβγ subunit is released and stimulates the muscarinic K+ channels to open. Hyperpolarization of the cardiomyocyte follows, dampening the contractile response. This is antagonized by RGS6
10.2.2.3 Gαq/11 Signaling
Angiotensin receptors (ATRs), endothelin receptors (ETRs), serotonin receptors and α-adrenergic receptors (αARs) are coupled to Gαq/11 signaling proteins [31]. Angiotensin I (AngI), Angiotensin II (AngII), and endothelin-1 (ET1) are examples of pro-hypertrophic hormones that bind these receptors during mechanical and/or pathological stress (i.e. stretch due to pressure or volume overload) [32]. Mechanistically, stimulation of Gαq/11-coupled receptors leads to activation of phospholipase C-β (PLCβ) and subsequent cleavage of membrane-bound phosphatidylinositol 4,5-biphosphate (PIP2) into the second messengers inositol (1,4,5) trisphosphate (IP3) and diacylglycerol (DAG) [33]. IP3 induces the IP3 receptor-mediated release of Ca2+ from the SR and is required to activate the downstream effectors CaMKII and calcineurin, whereas DAG diffuses along the plasma membrane and activates membrane bound forms of the serine/threonine protein kinase C (PKC) and/or protein kinase D, which in turn drive the downstream activation of ERK1/2 [33] (Fig. 10.4).
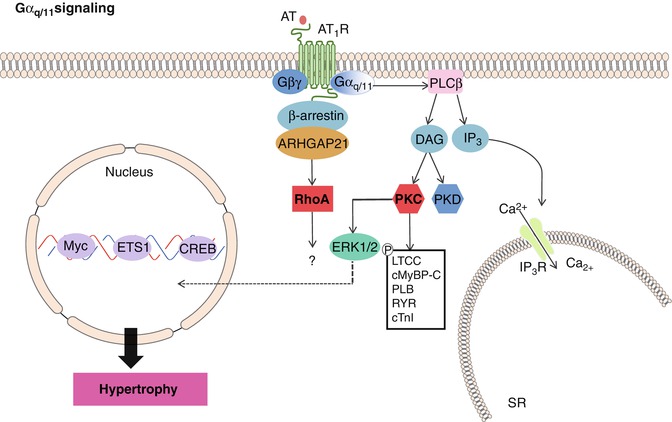
Fig. 10.4
Gαq/11 signaling. The AT1R is Gαq/11 coupled. Upon AT1R stimulation, PLCβ mediated generation of DAG and IP3 follows. IP3 induces IP3 receptor-mediated release of Ca2+ from the SR and is required to activate the downstream effectors CaMKII and calcineurin to drive hypertrophic transcription and induce cardiac contractility. DAG activates PKC and PKD. PKC leads to phosphorylation of LTCCs, cardiac myosin binding protein C, phospholamban, ryanodine receptor and cardiac troponin T, increasing cardiomyocyte contractility. In addition, increased ERK1/2 activity, leads to increased cellular transcription and cardiac hypertrophy results
Importantly, while all Gαq/11-coupled receptors function to control hypertrophy in response to cardiac stress, they each have additional important functional properties: α1-adrenergic receptor signaling is required to modulate Na+-H+ exchange transporter regulation; angiotensin receptor signaling controls Ca2+ signaling and collagen synthesis/deposition, and endothelin-1 mediates cardiac rhythm and collagen expression [34]. In addition, cardiac hypertrophy associated with Gαq signaling mediates the reactivation of embryonic genes, such as atrial natriuretic factor (ANF), skeletal α-actin, and βMHC [34].
Taken together, activation of Gαq signaling leads to an increase in transcriptional regulation of genes that control cardiomyocyte cell size and protein content, which over time, ultimately leads to hypertrophy and cardiac failure [35]. Indeed, transient overexpression of a constitutively active mutant of Gαq in transgenic mice consistently results in cardiac hypertrophy and dilatation with progressive heart failure and death [36]. Specifically, the transgenic overexpression of the angiotensin-1 (AT1R) receptor develops significant cardiac hypertrophy, with increased expression of atrial natriuretic factor (ANF) and interstitial collagen deposition, and the mice die prematurely of heart failure [37]. In contrast, mice carrying inactivating mutations of Gαq or Gα11 exhibit embryonic cardiomyocyte hypoplasia, suggesting that Gαq/Gα11-mediated signaling not only regulates myocardial growth during embryogenesis, but likely plays a critical role in pathological processes leading to cardiac hypertrophy and failure as well [38]. Importantly, moderate levels of Gαq signaling induce myocardial hypertrophy, while chronic and sustained activation caused cardiomyocyte death and HF through downstream activation of p38-MAPK and Janus kinases/signal transducer and activators of transcription (JAK/STAT) signaling pathways [39].
Therapeutically, it has been demonstrated that angiotensin-converting-enzyme inhibitors (ACEIs) are beneficial in hypertensive patients, as well as in patients with acute myocardial infarction (MI), chronic systolic HF, stroke and diabetic renal disease, specifically by targeting GPCR signaling [40]. Patients treated with ramipril, an ACEI, have improved clinical outcomes, with diminished left ventricular dysfunction, and reduced rates of death, myocardial infarction, and stroke [41]. Importantly, these data demonstrate a role for Ang II-mediated Gαq signaling in the development of cardiac hypertrophy and failure; however, the data also imply that the potential for targeted therapeutic interventions also exists, likely through specific inhibition of the Gαq downstream signaling effectors.
10.2.2.4 Gα12/13 Signaling
The most recently identified class of heterotrimeric G proteins is the Gα12/13 family. Gα13 was first identified as an oncogene, and overexpression of GTPase-deficient Gα12 or Gα13 altered cell shape, gene expression, and cell growth [42]. It is now clear that these responses resulted, at least in part, from activation of the small G protein Rho [43, 44] .
The Rho family of small G proteins includes more than 18 members, the best characterized of which are RhoA, Rac1, and Cdc42. A molecular link between the Gα12/13 family and RhoA was recently identified through work demonstrating that p115RhoGEF binds Gα12/13 proteins and is activated by Gα13 [43, 44] (Fig. 10.5). Similar interactions were shown for other GEFs, e.g., PDZRhoGEF and LARG, and Gα13 [42]. Interestingly, there is also evidence to suggest that signaling via AT1R-Gα12/13-mediated p115RhoGEF contributes specifically to activation of JNK-MAPK in response to both phenylephrine (PE) [45] and ET1 [46] (Fig. 10.5). Interestingly, GPCRs that couple to Gα12/13 also couple to (and signal through) other sub-classes, often Gαq/11.
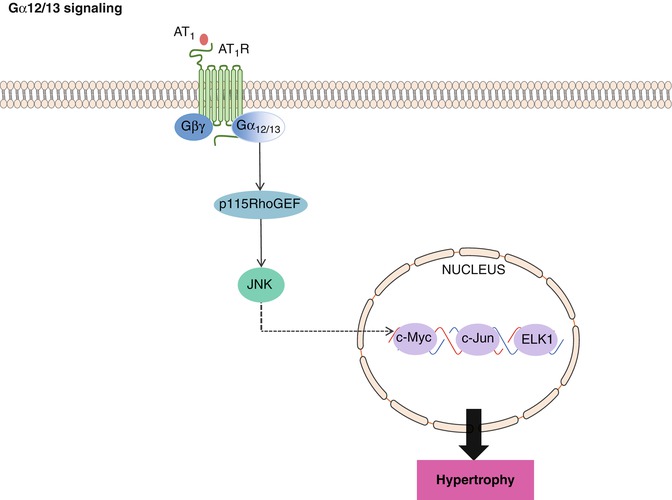
Fig. 10.5
Gα12/13 signaling. Stimulation of Gα12/13-coupled AT1R leads to p115RhoGEF mediated activation of JNK, which subsequently binds to nuclear transcription factors, increasing the expression of several hypertrophic genes, leading to cardiac muscle hypertrophy
10.2.2.5 β-Arrestins and Desensitization Signaling
Normally, the cell has mechanisms in place to turn GPCR signaling off. This inactivation can occur by one of two ways: The first is vesicular translocation, where the GPCR is engulfed by a clathrin-coated vesicle, removed from the membrane, disassembled from its agonist, and then is either recycled or degraded [47]. The second consequence is β-arrestin linking; here, the GPCR becomes phosphorylated by a G protein-coupled receptor kinase (GRK), recruiting β-arrestin and blocking further communication between the G protein and the GPCR agonist [48] (Figs. 10.2 and 10.4). This latter mechanism is termed desensitization [49]. In many cases, binding of β-arrestin to the receptor is a prerequisite for translocation. For example, β-arrestin bound to β2-ARS acts as an adaptor for binding with clathrin and with the beta-subunit of AP2 (clathrin adaptor molecules); thus, the arrestin here acts as a scaffold assembling the components needed for clathrin-mediated endocytosis of β2-adrenoreceptors [50].
Activity of GRKs and subcellular targeting is tightly regulated by interaction with receptor domains, G protein subunits, lipids, anchoring proteins and calcium-sensitive proteins [51]. In the myocardium, the β-adrenergic signaling exchange protein activated by cAMP (EPAC) plays a critical role in regulating cardiomyocyte handling and myofilament protein phosphorylation through activation of β-arrestin [52] (Fig. 10.2). Upon stimulation of β1AR in CMs, the scaffolding proteins β-arrestin 1 and β-arrestin 2 interact with both EPAC and CaMKII, allowing cAMP-EPAC-mediated activation of CaMKII and subsequent PLB phosphorylation and regulation of heart muscle contraction [30] . In addition, β-arrestin-dependent β1AR-mediated epidermal growth factor receptor (EGFR) transactivation decreases cardiac apoptosis, possibly via internalization of a β1AR-EGFR-ERK1/2 complex that directs an unknown cytosolic cell survival response. AT1R stimulation also induces β-arrestin-1 association with a Rho GTPase-activating protein called ARHGAP21, promoting RhoA activation and inducing changes in cytoskeletal structure and cell shape [53] (Fig. 10.4).
Interestingly, although various cardiac GPCRs have negative effects when stimulated chronically, β-arrestin has recently been recognized to mediate potentially beneficial downstream signaling. Internalization of an AT1R-β-arrestin-ERK1/2 complex has been shown to increase MAP kinase-interacting kinase-1 activation, enhancing eukaryotic translation of initiation factor-4E-mediated mRNA translation and contributing to the compensatory increase in cell size and protein content in response to hypertrophic stimuli. In addition, AT1R-β-arrestin-ERK1/2-mediated activation of ribosomal S6 kinase (rS6K) inhibits BAD-induced apoptosis, which could contribute to increased cardiomyocyte cell survival [30].
10.2.2.6 Calcineurin/NFAT Signaling
There is a clear correlation between force production and perturbation of Ca2+ regulation, alterations of which might directly induce pathological, anatomical, and functional alterations that lead to HF. Importantly, GPCR signaling leads to activation of PLC, inducing Ca2+ release from the sarcoplasmic reticulum (SR) which activates calmodulin, phosphorylates calcineurin, and, ultimately, modulates sarcomeric contractility and induces the transcription of hypertrophic genes (Fig. 10.6).
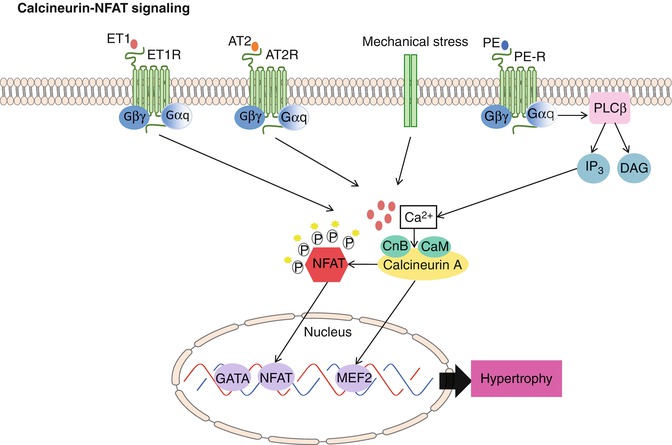
Fig. 10.6
Calcineurin/NFAT signaling. Upon stimulation of Gαq coupled receptors (i.e., ET1R, AT2R, or PE) or mechanical stress, the levels of intracellular calcium increase. Calcium then binds to calmodulin, thereby activating calcineurin through sustained elevations in intracellular calcium. This leads to nuclear localization of NFAT transcription factors as well as direct activation of nuclear MEF2 factors. Subsequent cardiac muscle hypertrophy follows
Calcineurin is a calcium- and calmodulin-dependent protein serine/threonine phosphatase critical for several important cellular processes [54]. It is expressed in multiple tissues and consists of two basic subunits: subunit A, the catalytic subunit; and subunit B, the regulatory subunit [54]. To date, there have been three mammalian calcineurin A catalytic subunit genes (a, b, c) and two B regulatory subunit genes (B1, B2) identified [55]. Importantly, calcineurin is uniquely activated by sustained elevations in intracellular Ca2+ [56].
Use of the immunosuppressive drugs cyclosporine A (CsA) and FK506, which form complexes with immunophilin protein and thus inhibit calcineurin catalytic activity, essentially have elucidated the physiological role for this phosphatase. Specifically, when cytoplasmic Ca2+ levels increase inside the cell, calmodulin associates with calcineurin, thereby activating the enzyme [17]. Consequently, cytosolic NFAT becomes dephosphorylated, allowing it to translocate to the nucleus where it can participate in mediating calcium-inducible gene expression [57] (Fig. 10.6).
To help modulate the signal, kinases such as ERK1/2, JNK, p38, glycogen synthase kinase-3β (GSK3β) and casein kinase I (CKI), casein kinase II (CKII), and PKA phosphorylate NFAT family members, antagonizing nuclear translocation [58]. In addition, many of the same kinases also participate in re-phosphorylating NFAT within the nucleus, aiding in extrusion of NFAT back into the cytosol [58]. Indeed, transgenic mice that overexpresses activated forms of calcineurin or NFAT3 in the heart develop cardiac hypertrophy that progresses rapidly to dilation, with interstitial fibrosis, congestive HF, and sudden death [59]. However, use of CsA and FK 506 block the ability of cultured CMs to undergo hypertrophy in response to AngII and PE stimulation [59]. Furthermore, CsA administration prevents cardiac hypertrophy and the associated pathology in calcineurin transgenic mice, suggesting calcineurin activation exacerbates hypertrophic signals and expedites the transition to dilation. Together, these results define a novel signaling pathway that couples hypertrophic signals at the cell membrane to changes in cardiac gene expression and suggests possible opportunities for pharmacologic intervention to prevent cardiac hypertrophy and HF [17].
Several endogenous calcineurin inhibitors exist, including AKAP79, Cabin/Cain-1, and DSCR/MCIP [60, 61]. Specifically, AKAP79 interacts with calcineurin, PKA, and PKC to serve as a scaffold that integrates these signaling pathways [62]. Cain/Cabin-1 is a 230-kDa protein highly expressed in the brain that contains a potent noncompetitive calcineurin inhibitory domain in its C terminus [60, 61]. The fact that neither AKAP79 nor Cain is expressed at significant levels in the heart suggests that they are not physiologic regulators of cardiac calcineurin activity [63]. On the other hand, MCIP1 and MCIP2 (DSCR1 and ZAKI-4) were recently identified calcineurin inhibitory genes shown to be highly expressed in the myocardium, potentially representing physiologic regulators of calcineurin activity in cardiac muscle [14].
10.2.2.7 MAPK Signaling
Nearly all MAPK signaling components (upstream and downstream) are activated in end-stage human HF and are subdivided into three main branches consisting of p38 kinases, JNKs, and ERK1/2 and ERK5 [15]. The MAPK signaling cascade is classically initiated by activation of small G proteins in response to both RTK-mediated and GPCR-mediated activation. However, the functional outcome of specific MAPK activation can be modulated and altered by scaffolds in a specific spatiotemporal pattern [64] (Fig. 10.7). This complexity leads to different phenotypes ranging from “physiological” compensated hypertrophy and cardioprotection to pathological HCM and cardiac remodeling, determined by the level, duration, mode, and timing of induction involving different isoforms and upstream/downstream pathways [64].
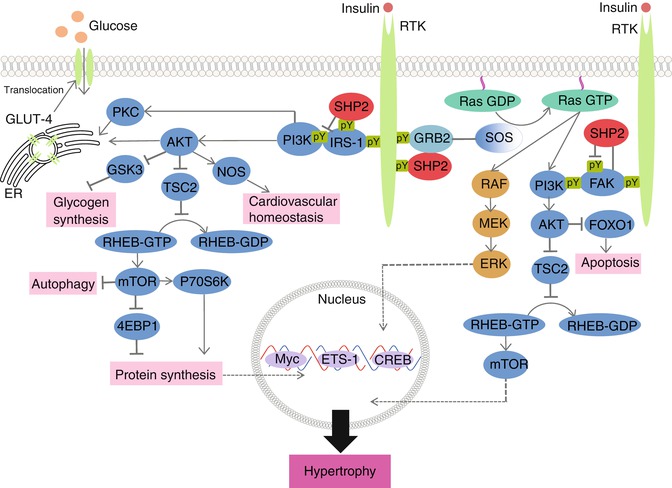
Fig. 10.7
Signaling pathways leading to SHP2-dependent signaling pathways leading to hypertrophy. When an RTK, such as insulin, binds to the receptor, both ERK/MAPK and PI3K/AKT signaling ensues. Importantly, ERK/MAPK translocates to the nucleus where it binds to transcription factors necessary to induce cardiac hypertrophy. The PI3K/AKT pathway will lead to the downstream activation of glycogen synthesis, autophagy and protein synthesis, culminating in mechanisms that can lead to cardiac hypertrophy. The PI3K pathway is also important in regulating the metabolic effects of insulin signaling. Stimulation of PKC and AKT by PI3K leads to translocation of GLUT-4 from the endoplasmic reticulum to the plasma membrane to mediate glucose metabolism in the cardiomyocyte
Pharmacologic inhibition of MAPK-signaling has demonstrated a necessary role for p38, JNK and ERK1/2 signaling in mediating hypertrophic growth of cultured CMs [65, 66]. In mice, constitutive activation of ERK1/2 signaling in the heart through expression of activated MEK1 causes a concentric type of hypertrophy [67]. In contrast, cardiac-specific inhibition of ERK1/2, through either a constitutively active ERK1/2-dual-specificity phosphatase or combinatorial deletion of ERK1 and ERK2, induces cardiac dilation [15]. Interestingly, and in contrast to ERK1/2, the related MEK5-ERK5 branch of the MAPK cascade preferentially induces eccentric hypertrophy [15]. Transgenic mice with cardiomyocyte-specific overexpression of MEK5, which induces downstream ERK5 activation, develop cardiac dilation and decompensation with loss of contractile function [68].
The JNK and p38 MAPK kinases, are activated by MEK4/7 and MEK3/6, respectively. These MAPKs generally serve as more specialized transducers of stress or injury responses, hence their classification as stress-activated protein kinases. Overexpression of MKK6 (p38 activation) or MKK7 (JNK activation) in hearts of transgenic mice show severe cardiac dilation and HF [65, 66].
10.2.2.8 PI3K/AKT Signaling
Phosphatidylinositol kinases are phospho-lipids tethered at cellular membranes that contribute to the recruitment and activation of multiple signaling components. They play a particularly key role in cell survival pathways, in the regulation of gene expression and cell metabolism, as well as in the process of cell growth and differentiation [69]. PI3K activation via protein kinase C-dependent or cAMP-dependent pathways are essential for the hypertrophic growth of adult CMs [70] (Fig. 10.7). RTK stimulation by growth factors, such as the insulin-like growth factor-1 (IGF-1) and the platelet derived growth factor (PDGF), lead to activation of the PI3K signaling cascade [70]. In addition, GPCR activation of both α-adrenergic and β-adrenergic signaling can also activate this pathway [70, 71].
Akt is the major downstream effector of PI3K signaling, mediating many processes important to cardiac adaptation including protein synthesis, inhibition of apoptosis, and metabolism. For example, cardiomyocyte-specific overexpression of the constitutively active catalytic subunit of PI3K, p110α, induces physiological, not pathological, H [72]. Conversely, dominant negative p110α overexpression in the heart induces a non-pathological atrophy, even in the presence of Insulin-like growth factor 1 receptor (IGFR) overexpression and exercise training [72]. Cardiac-restricted loss of the lipid phosphatase, phosphatase and tensin homolog deleted on chromosome ten (PTEN), which increases phosphorylation of Akt and GSK-3β, also promotes heart growth and prevents the development of maladaptive ventricular remodeling, with preservation of angiogenesis and metabolic gene expression in response to pressure overload [73].
Akt1-null mice, however, display growth retardation and are refractory to physiological cardiac hypertrophy when subjected to exercise training. Similarly, overexpression of dominant negative Akt1 prevents hypertrophic growth of the heart [74]. Importantly, while acute cardiomyocyte-specific expression of a constitutively active Akt1 initially promotes a physiological type of hypertrophy, its prolonged activation is ultimately pathological [74, 75]. Taken together, these results indicate that acute Akt activation promotes an adaptive cellular growth program in the heart, but that sustained Akt signaling leads to pathological hypertrophy and heart failure [76]. Indeed, the mediators of PI3K/Akt-induced hypertrophy are two well-defined direct downstream targets: GSK3β and the mammalian target of rapamycin (mTOR) [14].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


