Fig. 17.1
Electron micrograph of an isolated adult cardiocyte
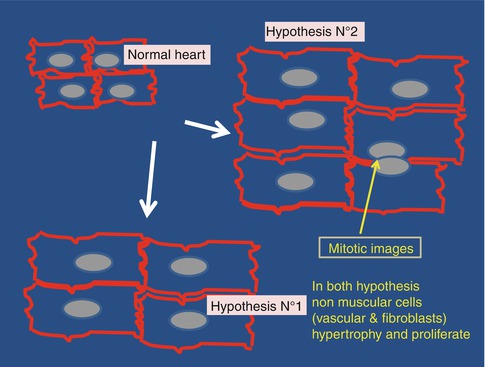
A second thesis was supported since a long time by the group of Piero Anversa [12] (Fig. 17.2). According to Anversa cardiocytes remain able to multiply during all their life and are still capable to regenerate, even in old persons. In addition, there are a substantial amount of stem cells within the heart and these stem cells remain active in adult and even in aged hearts, which may have practical consequences for regenerative surgery. A technological revolution has recently bring strong support to this view, the retrospective C14 cellular birth dating. Surprisingly such a revolution was due to the Cold War and the nuclear bomb [13, 14]. Before the Cold War the atmospheric radioactive C14 concentration was very low and stable. In 1963, the occurrence of nuclear assays acutely enhanced this concentration. In 1970, nuclear assays stopped which results in an exponential decay in atmospheric concentration in the isotope. C14 is found in every tissue of individuals born after this date [13] and it is therefore possible to quantitate the turn-over of a given category of cells providing the method to quantitate the radioactive compound is sensitive enough. The group of Anversa was able to measure the turn-over of cardiocytes on human hearts and found that, approximately, the age of these cardiocytes was 8 months in children, 8 years in adults and 2 years and a half in aged persons. The conclusion was that the turn-over rate of aged cardiocytes is accelerated (probably by the increased aortic impedance that causes an impedance mechanical overload in aged hearts, see below [15]). In addition, they found that, between the age of 20 and 80 years cardiocytes, EC and fibroblasts of the myocardium were respectively replaced 8, 6 and 8 times. Mechanical overload enhances this turn-over rate.
Cardiocyte HYP is the result of a sarcomeric reorganisation. Dilation of the heart is associated with myocyte lengthening mediated by the generation of new sarcomeres in series. The result is a pronounced enhancement of the length/width ratio of the myocytes. The same phenomenon is observed in the rat 2 or 6 days after birth. This would suggest that during CR a foetal gene program involving activation of the cytoskeletal proteins is initiated which regulates the overall shape of the myocyte by preferentially directing lengthening of the cell. In contrast, HYP of cardiocytes is the result of the addition of new sarcomeres in parallel. Such a hypothesis is favoured by the observation made by J.-L. Samuel et al. showing a reversible rearrangement of the microtubular network during the early stage of cardiac overload [16].
17.2.1 Anatomical Remodeling
The current belief that the heart is a sphere or a rugby balloon, is against evidence. The human heart and the bird heart, are more like a mop and hypertrophy or dilatation are not simply an exaggeration of the balloon structure but have also modified the structure of the mop. Ventricles do not contract homogeneously; during contraction the apex does not move despite the fact that it is free, while the basis, that is attached, is moving periodically up and down. The diastolic filling cannot be explained using the balloon image and requires a model in which filling is an active process and this is the case in such a model: In Spain, an anatomist named F. Torrent-Guasp provided the response by showing that in fact the ventricular myocardium is constituted by a single contractile band that is submitted during embryogenesis to several modifications. The final result is a double helix structure of the ventricles [17, 18]. It is possible to quantify the torsion of the heart [19], but such a measure is not routinely performed despite the fact that this likely to provide a better estimation of myocardial performance than the classical ejection fraction.
The contractile band is unique and begins below the pulmonary artery and then takes a rightward direction to form the right ventricle, the posterior wall of the left ventricle, followed by the descending and the ascending fibers of the left ventricle to end as an apical loop; the subepicardium becomes subendocardium during the formation of the loop [17, 20]. CR strongly modifies the “mop” structure of the heart, especially during dilatation. Fibers progressively lose their helicoidal shape by becoming parallel which per se will seriously impair the contractile performance. The mop effect is also strongly attenuated by fibrotic bands and the loss of homogeneity [17].
The increase in left ventricular (LV) mass and volume is accompanied by a change in the shape of the ventricle. If the process is triggered by a MI, the remodelling is asymmetric and is associated with infarct expansion. Early ventricular remodelling following coronary occlusion is dominated by infarct expansion which is an acute dilation caused by death and slippage of the myocytes; it dramatically alters ventricular volume and geometry.
Ventricular remodelling after MI is the result of infarct expansion and volume-overload hypertrophy of non-infarcted myocardium. Later on CR is associated to additional enlargement and sphericity of the ventricle, a decrease in stroke volume and impaired diastolic filling [this aspect of remodeling has been developed in 7]. Systolic impairment secondary to the loss of contractile material results in an increased end-systolic volume, an increased cardiac size and a secondary augmentation of the diastolic filling pressure and distensibility. As the fibrosis increases, the distensibility decreases resulting in an increase in diastolic pressure and volume. Peripheral mechanisms including vasoconstriction subsequently increase both preload and afterload; this produces an increased wall stress and a progressive thinning of the area. Simultaneously in non-infarcted segments, elevation of the end-diastolic stress causes volume-overload HYP which tends to normalise the wall stress according to Laplace’s law. The extent and location of the infarction, therapy or associated diseases, may considerably modify remodelling [21]. Ventricular HYP remains symmetric and concentric when the cause of CR is pressure overload due to arterial hypertension or aortic stenosis, by contrast volume overload creates dilatation and renders the fibers more parallel and attenuates the loop structure [7].
17.2.1.1 Physiological Mechanisms of Adaptation
In every organ or organism, the so-called “phenotypic plasticity” is a general mechanism widely utilized in nature to adapt organs to new environmental conditions [8, 22]. In muscle physiology, biological adaptation is an evolutionary process by which an organ structure, i.e. the structure of a muscle is modified to obtain a thermodynamic status adapted to the new mechanical conditions. Skeletal or cardiac muscles are equally “plastic” and equally capable to adapt to changes in chronic loading conditions by both increasing the contractile mass and modifying their maximum shortening velocity. In terms of economy, the myocardium is slightly continuously different from the skeletal muscle: (i) it has indeed to contract day and night and then requires a highly efficient oxygen-dependent source of energy as mitochondria; (ii) it has a more or less “spheric shape” as already described, and as such respond to the Laplace law (Fig. 17.3).
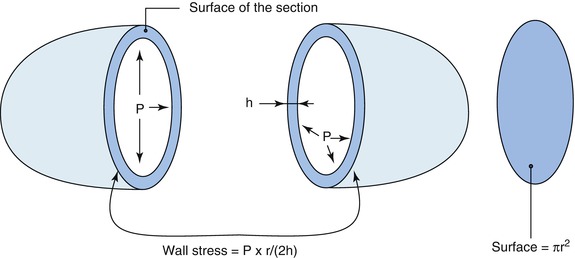
Fig. 17.3
The Laplace law: dilatation of the heart requires hypertrophy to maintain a normal wall stress
In the heart, changing loading conditions, for example during pressure overload after aortic stenosis, is just like increasing the load on a skeletal muscle fiber (or the load on a car). To maintain contraction (or, for the car, to pursue the trip), the cardiac contraction velocity becomes slower (the driver has to slow his car), and, in the same time, the muscle economy (the amount of work per mol of Adenosine Tri-Phosphate, ATP) drops [5].
This adaptation process begins very rapidly and is dominated by several changes in genetic expression which allow the heart to recover a normal economy and to maintain a normal contraction for a long period of time. Quantitative modifications lead to cardiac hypertrophy, and, according to Laplace’s law, will normalise the wall stress (Fig. 17.3). Simultaneously, molecular changes will allow the myocardial fibre to contract more slowly and to recover a normal economy by having a lower maximum shortening velocity, Vmax (see further) [5]. The permanent changes in genetic expression simultaneously have, even at the beginning, both beneficial effects in terms of muscular economy and detrimental, or even useless, consequences. Heart Failure, (HF), when it occurs, indicates the limits of the process of biological adaptation.
Such a change in the phenotype is the same in a variety of conditions, including foetal development, volume and pressure-overload, senescence or hypothyroidism. The same programme is always used simply because this is the only developmental program available in the heart; (in contrast, in the skeletal muscle, two programs are available, a foetal and an embryonic program, they both are re-expressed during mechanical overload [23]).
17.2.2 Cardiac Hypertrophy
Cardiac HYP enables the heart to adapt the new working conditions by both multiplying the contractile units and reducing the wall stress according to Laplace’s law. The activation of protein synthesis is a rapid and rather homogeneous phenomenon, it belongs the foetal reprogramming as well, and, for example, the first signs of an activated synthesis can be observed within half an hour after banding the aorta of a rabbit [4] (Tables 17.1 and 17.2). Others have shown that total protein, myosin, myoglobin, and collagen synthesis, were activated after 3 h of increased aortic pressure. The accumulation of total proteins or myosin is due to an increased rate of synthesis, nevertheless the rate of degradation is also augmented in parallel; such a paradoxical wasting effect is a general feature in protein metabolism [references in 6].
Table 17.1
Energy transmission in cardiac mechanical overload expressed in calories per gram (heat produced)
Type of heat | Origin | Variation in hypertrophy (%) | |
|---|---|---|---|
During contraction | Initial heat | Sliding process | −54a |
Ca movements | −60a | ||
After contraction | Recovery heat | Mitochondrial activity | −20 |
Permanent survival of the tissue | Resting heat | Ionic milieu and synthesis activity | −1 |
Changes in contractile activity | |||
Force (N/mm2) | Force-time integral | +17a | |
Shortening velocity | −48a | ||
Table 17.2
The fetal reprogramming in cardiac overload
Changes in gene expression | Consequences |
|---|---|
Global increase in gene expression | Hypertrophy and normalization of wall stress |
Activation of microRNAs (non coding) | Fine tuning of the foetal program |
Genes induced or re-expressed | |
Increased expression of beta myosin heavy chain, ANF and BNP, neuronal NO synthase and caveoline; anaerobic switch (LDH-M, CK-B) | Decreased myosin ATPase and Vmax; decreased exercise performances, better recovery period Increased Economy |
Increased alpha-3 subunit of Na-K-ATPase | Decreased Na affinity |
Increased genes coding the apoptotic pathway | “Eat-me” signal occurs at the early beginning after an overload |
Genes expression blunted (the concentration of the corresponding protein diminishes) | |
Ca-ATPase of SR (SERCA2), early transient K current It0, alpha myosin heavy chain; | Increased action potential duration and relaxation time |
Beta-1 adrenergic and muscarinic receptor | Decreased contractility during exercise |
Myoglobin | Participates in the anaerobic switch |
17.2.2.1 Thermodynamic Adaptation
In order to quantitate the economy of a system, it is necessary to measure the mechanical performance indices such as force, or force/time integrals, or work (economy is then termed efficiency) and the corresponding energy flux. The energy flux can be quantitated by measuring ATP, or oxygen consumption, or heat production (the heat production is an index of economy according to the second Carnot principle in thermodynamic, see [5] for further explanations). A major technical progress was made following the invention of a microthermopile which allowed N. Alpert’s group to measure heat production on a rabbit papillary muscle as well as in strips of human cardiac muscle on a beat-to-beat basis.
The heart, as any other muscle, utilizes energy: (i) for the simple survival of the tissue, such as protein synthesis and ion movements (“resting heat”); (ii) resynthesis of the high-energy phosphate stores which mainly occurs in mitochondria, but can depend on the anaerobic glycolytic pathway during ischemia (“recovery heat”); (iii) contraction (“initial heat”), including ATP hydrolysis for cross-bridge cycling (“tension-dependent initial heat”), and excitation-contraction coupling (“tension-independent initial heat”) (Table 17.1).
The fundamental process of adaptation which occurs during mechanical overload, both in human and in experimental models, includes a slowing of Vmax with a diminution of the heat produced per g of active tension during contraction. Both the resting and recovery heats remain unchanged. Special experimental protocols allow one to partition heat produced by the sliding process, from that produced by the movement of calcium and the activity of the different calcium pumps. Both systems are used during contraction and both function more economically during cardiac HYP in animal models and in end-stage HF in humans [5].
To conclude: (i) the reduction in Vmax is the main basic process responsible for myocardial adaptation to mechanical overload. Thermodynamic data has suggest that this reduction is due to a decreased recruitment of myosin cross-bridges. Such modifications already exist in the compensated stage. (ii) In sharp contrast with the current bedside opinion, the diminution of Vmax (the initial shortening velocity, which is also the maximum shortening velocity and is obtained by extrapolation of the shortening velocity/load curve) has a beneficial event at least at the myofiber level, since it allows the cardiac fiber to contract at a normal energy cost. Nevertheless at the organ level the diminution of Vmax is also the first step which will finally lead to a decrease in cardiac output and finally to failure. (iii) Perturbed mitochondrial oxidative phosphorylation or anaerobic energy metabolism are both unlikely candidates to cause HF since the “recovery heat” remains unchanged (despite a non statistically significant 20 % decrease) even during end-stage HF in dilated cardiomyopathy. Hence, investigations concerning the adaptational process have to focus on energy utilisation rather than energy production (Table 17.1).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


