Pathophysiology of Ventricular Dyssynchrony and Mechanisms of Cardiac Resynchronization Therapy
Robert H. Helm
David D. Spragg
Khalid Chakir
David A. Kass
HEART FAILURE AND RESYNCHRONIZATION THERAPY
Heart failure (HF) is a systemic disease, which typically begins with an initial cardiac insult resulting in acute ventricular dysfunction. To maintain pump function and systemic perfusion, a series of neurohormonal and adrenergic adaptations occur. Chronically, these changes lead to remodeling and maladaptation at the molecular and cellular levels resulting in sustained pump dysfunction.1, 2 Neurohumoral stimulation occurs at both the systemic and local myocardial levels, and myocardial responses can be induced by hormonereceptor interactions, or mechanical stress.3 Catecholamines acutely recruit myocardial reserve; however sustained stimulation results in abnormal calcium handling and worsened myocyte survival.4 Intersitial fibrosis, vascular insufficiency, activation of fibroblasts and matrix remodeling protein (metalloproteinase), and other factors contribute to chronic maladaptive remodeling.1 The result is impaired basal and reserve heart function manifested by blunted responses to neurohumoral stimulation (i.e., sympathetic stimulation), loading, and increases in heart rate. Many factors contribute to this behavior, including down-regulation of G-coupled receptor pathways, abnormal calcium cycling,5,6 activation of multiple stress response signaling pathways,7,8 energetics,9 and sarcomere changes. Impaired Ca2+ cycling into and out of the sarcoplasmic reticulum blunts myocardial force generation and delays relaxation. Activation of stress kinases, phosphatases, and associated transcription factors alters contractile function, calcium handling, growth remodeling, and cell survival. Cardiac bio-energetics is rendered inefficient with depressed fatty acid utilization. Lastly, myofilament proteins are altered in the failure state, including the type of myosin, and post-translational modifications of regulatory and structural proteins such as titin.1,10 These and other pathologic alterations all contribute to the HF phenotype.1
On top of this disease landscape, about one-third of patients develop electrical conduction delays resulting in marked discoordinate contraction.11,12 Dyssynchrony reduces chamber function and efficiency in a normal heart, and its superimposition in HF worsens an already compromised state. Furthermore, dyssynchrony effects may not be simply additive; they may trigger more complex pathophysiology and a unique form of heart failure. Understanding how and what is achieved by CRT is increasingly important, as it has become standard therapy for groups of heart failure patients. In this chapter, we update current understanding of the mechanical, cellular, and molecular changes induced by dyssynchrony in the failing heart and how CRT affects them.
Mechanics of Dyssynchrony
His-Purkinje disease and intra myocardial conduction delay in the failing heart result in regionally delayed electrical excitation,13 yielding early- and late-activated regions that contract out of phase with each other to generate dyssynchrony. Both left-bundle branch block (LBBB) and right ventricular (RV) pacing delay lateral wall stiffening and are the most common causes of left ventricular (LV) dyssynchrony. With both, septal activation occurs first, but as the lateral wall remains quiescent, forces from septal contraction do not raise LV pressure but are largely converted into prestretch of the lateral wall. This slows the rise of pressure (dP/dt) and increases lateral wall stress. When the free wall does contract, it generates systolic forces late, which are partly dissipated by restretching the already relaxing septal region, lowering net cardiac output. Delayed papillary muscle activation can contribute to mitral regurgitation, further reducing forward ejection. This common pattern of mechanical dyssynchrony is shown in Figure 1.1A. Tagged magnetic resonance imaging (MRI) was used to assess regional strain in a canine with mechanical dyssynchrony induced by radiofrequency ablation of the left bundle.14 Strain is plotted as a function of LV region. In early systole (dashed line), the septum contracts (negative strain), whereas the lateral wall stretches (positive strain). In late systole (solid line), this pattern is reversed; as the lateral wall contracts, the septum stretches. Regional disparities in cardiac stiffening can be appreciated by portraying dyssynchrony as two time-varying elastance curves representing the early- and late-activated regions (Fig. 1.1B). The vertical distance between curves reflects disparities in wall stiffening that generate discoordinate motion. This is most marked in early
systole (isovolumic contraction, lowering dP/dtmax), and late systole as one territory enters relaxation ahead of the other. The latter is when echoDoppler measures of dyssynchrony are typically observed.15,16 The evolution and resolution of dyssynchrony highlight a critical time course and provide insight into how delayed contraction affects cardiac function throughout the cardiac cycle.
systole (isovolumic contraction, lowering dP/dtmax), and late systole as one territory enters relaxation ahead of the other. The latter is when echoDoppler measures of dyssynchrony are typically observed.15,16 The evolution and resolution of dyssynchrony highlight a critical time course and provide insight into how delayed contraction affects cardiac function throughout the cardiac cycle.
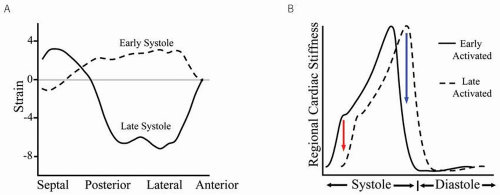 FIG. 1.1. A: Circumferential strain (relative shortening) at different regions across a short-axis section of the mid-LV in a dyssynchronous heart. Data for early (dashed) and late (solid) systole are shown, and reveal septal and lateral regions out of phase with each other. B: Model of dyssynchrony based on a time-delay of ventricular activation (stiffening). Plots of early-(solid) and late-(dashed) activated myocardial regions. Vertical distance between the curves indicates transfer of forces from one region to the other, and the arrows highlight two times—early contraction and late systole where this disparity is greatest and where discoordinate motion most manifests. (A, adapted from Byrne MJ, Helm, RH, Daya, S, et al. Diminished left ventricular dyssynchrony and impact of resynchronization in failing hearts with right versus left bundle branch block. J Am Coll Cardiol. 2007;50:1484-1490.) |
In contrast to LBBB and RV-paced associated dyssynchrony, right-bundle branch block (RBBB) results in delayed right-sided activation relative to LV free wall. The impact of RBBB on chamber synchrony and function is significantly less than with LBBB14 largely due to the lack of symmetric heart geometry. Unlike the LV free wall, the septum is loaded not just by regional LV forces but also those from the RV. The size of the delay-activated region and its location are key factors that determine the net impact on global LV function and mechanical synchrony.
Dyssynchrony, Relaxation, and Loading
Cardiac dyssynchrony can delay ventricular relaxation. In an acute pacing study in dogs, Aoyagi et al.17 demonstrated a significant correlation between left ventricular dyssynchrony (measured in early diastole) induced with ventricular pacing and prolongation in relaxation (increase in Tau, time constant of relaxation, Fig. 1.2A). Increased LV afterload can also induce relaxation delay and dyssynchrony. With acute aortic constriction in an otherwise normal canine, Yano et al.18 showed an acute increase in Tau and onset of regionally delayed contraction (Fig 1.2B and Fig. 1.2C). Nearly 15 years later, attention is principally focused on cardiac dyssynchrony due to conduction delay (typically LBB pattern) but certainly abnormal loading associated with heart failure can contribute to chamber dyssynchrony. Wang et al.19 further substantiated these data by showing that vasodilators and diuretics could improve dyssynchrony (measured during the diastolic period) and relaxation in HF patients (Fig. 1.2D). These data are particularly important because dyssynchrony due to abnormal loading is far less likely to be amenable to electrical resynchronization.20
Effect of Dyssynchrony on Pump Function and Efficiency
One critical consequence of LV dyssynchrony is reduced pump function, which can be demonstrated by pressurevolume loops.21 Figure 1.3A shows example relations, and with dysynchrony induced by RV pacing, the end-systolic pressure-volume relationship (ESPVR) shifts rightward, indicating a fall in net function. Both stroke volume (loop width) and stroke work (loop area) also decline without a fall in end-diastolic volume. Furthermore, end-systolic wall stress is increased as the end-systolic volume rises. Rate of pressure development (dP/dtmax) declines by ˜20%, stroke work by 10-15%, and relaxation (Tau) prolongs by ˜10-15%.22,23
A second critical consequence of dyssynchrony is that LV efficiency is reduced. Work performed by one region of the heart is wasted by means of stretching the contralateral wall (intracavity energy sink). In a canine model of dyssynchrony induced with RV pacing, Prinzen et al.24 assessed regional myocardial strain and work using tagged MRI. At baseline (right atrial pacing) regional work was fairly homogeneous; whereas with RV pacing a marked disparity in local work including a 125% increase in the lateral wall and a reciprocal
decrease in the early-activated septum was observed. Local stress-strain plots illustrate these findings (Fig. 1.3B). Early shortening under low external load and late-systolic stretching under higher load creates a “figure-8” shaped stress-strain plot. The workload (loop area) for this territory is consequently low. In contrast, in the late-activated lateral wall, which functions at higher initial stretch and contracts against higher stress, the workload is much greater. These regional differences in work correlate with regional blood flow and metabolic demands.25,26,27 Total myocardial oxygen consumption (MVO2) is unaltered despite a striking decline in LV stroke work correlating with a marked decline in LV efficiency (work / MVO2 consumption).28, 29, 30
decrease in the early-activated septum was observed. Local stress-strain plots illustrate these findings (Fig. 1.3B). Early shortening under low external load and late-systolic stretching under higher load creates a “figure-8” shaped stress-strain plot. The workload (loop area) for this territory is consequently low. In contrast, in the late-activated lateral wall, which functions at higher initial stretch and contracts against higher stress, the workload is much greater. These regional differences in work correlate with regional blood flow and metabolic demands.25,26,27 Total myocardial oxygen consumption (MVO2) is unaltered despite a striking decline in LV stroke work correlating with a marked decline in LV efficiency (work / MVO2 consumption).28, 29, 30
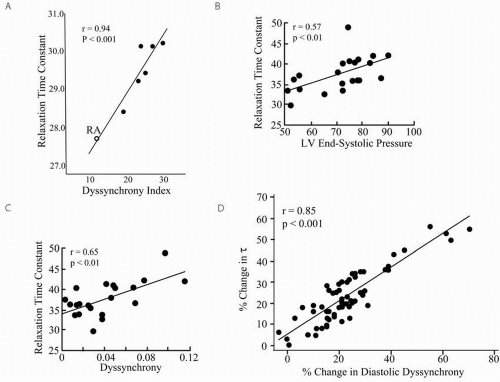 FIG. 1.2. A: Effect of mechanical dyssynchrony induced with ventricular pacing (closed-circles) on relaxation in a normal canine as compared with baseline synchronous contraction (right atrial pacing, open circle). Ventricular pacing was performed from various LV and RV sites. B and C: Cardiac dyssynchrony and delay in chamber relaxation induced by increasing afterload (systolic end-systolic pressure) by aortic occlusion. D: Percent change in relaxation and diastolic dyssynchrony after improving cardiac loading with pharmacologic therapy. (A, Adapted from Aoyagi T, Iizuka M, Takahashi T, et al. Wall motion asynchrony prolongs time constant of left ventricular relaxation. Am J Physiol. 1989;257:H883-H890. B and C, adapted from Yano M, Kohno M, Konishi M, et al. Influence of left ventricular regional nonuniformity on afterload-dependent relaxation in intact dogs. Am J Physiol. 1994;267:H148-H154. D, adapted from Wang J, Kurrelmeyer KM, Torre-Amione G, et al. Systolic and diastolic dyssynchrony in patients with diastolic heart failure and the effect of medical therapy. J Am Coll Cardiol. 2007;49:88-96.) |
Gross Pathologic Changes Associated with LV Dyssynchrony
Chronic dyssynchrony leads to maladaptive ventricular remodeling. Vernooy et al.26 assessed structural and functional changes 16 weeks after ablating the left-bundle branch in dogs to create cardiac dyssynchrony. A decline in LV EF (-25%) paralleled an increase in chamber diameter by +23%. As previously discussed, RV pacing also generates LV dyssynchrony, and Thambo et al.31 studied the long-term effects of RV pacing at physiologic heart rates in 23 patients with congenital heart block undergoing pacer implantation. Combined thinning of the early-activated septum and thickening in
late-activated posterior wall resulted in a 30% increase in the ratio of posterior to septal wall thickness. In addition, LV chamber diameter increased by 20%.
late-activated posterior wall resulted in a 30% increase in the ratio of posterior to septal wall thickness. In addition, LV chamber diameter increased by 20%.
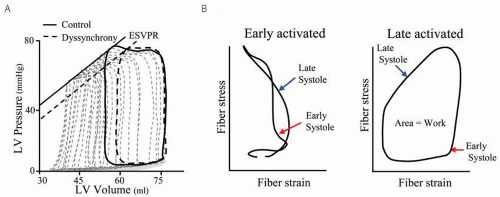 FIG. 1.3. A: Pressure-volume loops showing effect of dyssynchrony on end-systolic pressure-volume relation (ESPVR) and resting cardiac cycle (loop). The ESPVR shifts rightward, end-systolic volume increases, and stroke volume and work declines. B: Stress-strain loops from early- versus late-activated regions in a dyssynchronous heart. Whereas these loops would normally appear the same, with dyssynchrony the early-activated region first contracts at low load and is then stretched generating a figure-8 shaped loop with little area (reduced work). The late contracting lateral wall operates at higher preload and stress, requiring greater work. (A, adapted from Park, RC, Little WC, O’Rourke RA. Effect of alteration of left ventricular activation sequence on the left ventricular end-systolic pressure volume relation in closed-chest dogs. Circ Res. 1985;57:706-717.) |
Dyssynchrony induces regional changes in underlying cardiac fiber architecture. In a canine model of dysynchronous heart failure (LBB ablation followed by 3 weeks of atrial tachypacing), Helm et al.32 used high-resolution MRI and computational anatomical registering to show that, while the primary fiber orientation (epicardium downward, endocardium upward, and mid-myocardial circumferential) was not significantly altered, the transmural fiber gradient was markedly increased in the septal region owing largely to wall thinning. In addition, they found that the orientation of laminar sheets (planes of muscle fibers that constitute the myocardium) were oriented more vertically in the earlyactivated septum but not changed in the lateral wall. Such regional alterations in fiber architecture could affect the local biophysical/mechanical properties of tissue and impact propagation of electrical excitation.
Biochemical Consequences of Dyssynchrony
The combined effect of dyssynchrony and heart failure alters expression and activity of various proteins beyond that observed with heart failure alone. In the first study to test for such effects, Spragg et al.33 contrasted regional molecular changes in failing canine hearts with and without dyssynchrony (eg, right ventricular versus right atrial tachypacing). Sarcoplasmic reticulum (SR) Ca2+-ATPase, which actively transfers Ca2+ from the cytosol into the SR, and phospholamban, a coregulator of the latter protein, were down-regulated (˜20-30%) in the lateral endocardium versus other territories.
The extracellular response kinase (ERK1/2), a mitogen-activated kinase that is associated with stress stimulation pathways modulating cell survival and differentiation, was highly activated in the lateral endocardium versus other regions. Connexin 43 (Cx43), a gap-junction protein that allows for rapid cell-to-cell depolarization, was marked downregulated. Importantly, these molecular changes in the lateral endocardium were not observed in failing myocardium without dyssynchrony. Recently Chakir et al. 34 showed differential activation of stress response proteins, such as tumor necrosis factor-alpha, Ca2+-calmodulin-dependent kinase II, and p38 MAP kinase. Interestingly, these changes were only observed when dyssynchrony was combined with heart failure and not with dyssynchrony alone35—indicating that dyssynchrony interacts specifically with underlying heart failure substrate to trigger molecular alterations. Finally, Bilchick et al.36 similarly observed regional expression of genes involved with growth and hypertrophy, stress signaling, and matrix remodeling in normal mice after only 7 to 10 days of RV (dyssynchrony) pacing. Regional molecular polarization in the dyssynchronous failing heart may contribute to heterogeneous electromechanical coupling and enhanced arrhythmia susceptibility.
Dyssynchrony Alters Regional Electrical Heterogeneity and Arrhythmia Susceptibility
Heart failure and QRS prolongation11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37 are powerful and independent predictors of mortality due to ventricular arrhythmias in heart failure patients. Two prerequisites for reentrant arrhythmia are 1) unidirectional conduction block and 2) either sufficiently long circuit path-length or regionally delayed conduction allowing for adequate time to regain excitability
in the blocked limb. Selective prolongation of APD within mid-myocardial cells due to remodeling of delayed rectifier K current and possibly Ca2+ currents have been implicated in causes for QT-interval prolongation, transmural heterogeneity of repolarization, and susceptibility to conduction block (the first prerequisite for arrhythmia induction) observed in dyssynchronous HF.38,39 In addition, dyssynchrony alone (without HF) alters regional myocardial conduction and has been implicated in arrhythmia susceptibility, fulfilling the second prerequisite for reentry. Myocardial conduction is anisotropic with more rapid cell-to-cell conduction occurring along the myocardial fiber direction owing to gap junction protein coupling at intercalated discs. Spragg et al.35



in the blocked limb. Selective prolongation of APD within mid-myocardial cells due to remodeling of delayed rectifier K current and possibly Ca2+ currents have been implicated in causes for QT-interval prolongation, transmural heterogeneity of repolarization, and susceptibility to conduction block (the first prerequisite for arrhythmia induction) observed in dyssynchronous HF.38,39 In addition, dyssynchrony alone (without HF) alters regional myocardial conduction and has been implicated in arrhythmia susceptibility, fulfilling the second prerequisite for reentry. Myocardial conduction is anisotropic with more rapid cell-to-cell conduction occurring along the myocardial fiber direction owing to gap junction protein coupling at intercalated discs. Spragg et al.35
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
