Age category (years)
1988–1991 (%)
1999–2000 (%)
8–17
7.3a
9.7b
18–39
5.1
7.2
40–59
27
30.1
>60
57–9
65.4
31.2 Pathophysiology of Hypertension in Children and Adolescents
Hypertension in childhood is defined as a BP above the 95th percentile taking into account age, sex, and height [6]. The prevalence of hypertension in children and adolescents is generally well below 10 % in the population, but the exact figure depends very much on the conditions in which BP is measured and on the number of visits [7]. Thus, in a recent survey of a large group of schoolchildren, we found that the prevalence of hypertension decreased from 11 to 2.2 % between the first and the third visits. The prevalence of elevated BP in children and adolescents is associated with an excess of body weight, a high heart rate, and the family history of hypertension. However, the precise role of obesity in the recent increase in the prevalence of hypertension in childhood is debated [8].
In small children (<6–7 years), the detection of hypertension is commonly associated with the presence of a disease such as congenital renal abnormalities, a renal parenchymal disease, a coarctation of the aorta, or eventually a renal artery stenosis, endocrine diseases, and more rarely genetic alterations leading to hypertension [9] (Table 31.2). Hence, the pathophysiology of hypertension is linked directly to the mechanisms associated with the type of secondary hypertension. In congenital renal malformations or parenchymal renal diseases, the reduced glomerular filtration leading to a diminished capacity to excrete water and sodium plays an important role in the development of hypertension together with an activation of the renin-angiotensin and sympathetic nervous systems. In renal artery stenosis, the stimulation of the renin-angiotensin system by the stenotic renal artery contributes to BP elevation in order to maintain sodium balance at the expense of a high systemic BP. In young patients, a coarctation of the aorta induces hypertension through a dysfunction of baroreceptors associated with an intimal and medial hypertrophy of large and small vessels and a stiffening of the aorta. These changes can induce a vascular endothelial dysfunction, a reduced vasodilatory response to acetylcholine, and an increased vasoconstrictive response of peripheral vessels to catecholamines. For these reasons, hypertension tends to persist after correction of the coarctation.
Table 31.2
Common causes of hypertension in children between 1 and 10 years
First year | Coarctation of the aorta |
Renovascular disease | |
Renal parenchymal disease | |
Iatrogenic (medication, volume) | |
Tumor | |
Infancy to 6 years | Renal parenchymal disease |
Renovascular disease | |
Coarctation of the aorta | |
Tumor | |
Endocrine causes | |
Iatrogenic | |
Essential hypertension | |
Age 6–10 years | Renal parenchymal disease |
Essential hypertension | |
Renovascular disease | |
Coarctation of the aorta | |
Endocrine causes | |
Tumor | |
Iatrogenic |
The pathophysiology of endocrine-mediated hypertension depends on the type of hormone overproduction or absence. Glucocorticoid- and mineralocorticoid-induced hypertensions are mediated primarily by the sodium- and water-retention properties of these steroids. In addition, direct effects of these hormones on cardiac, vascular, and renal tissues participate in the elevation of systemic BP. For example, glucocorticoids increase the synthesis of angiotensinogen and reduce the synthesis of prostacyclin resulting in a greater vascular reactivity to angiotensin II and catecholamines. Aldosterone has well-described profibrotic effects in the heart, kidneys, and vessels [10]. Hypertension is also a clinical characteristic of acromegaly. In that situation, hypertension is the consequence of the excess of growth hormones which leads to vascular hypertrophy but also to a hyperdynamic state, due to an activation of the sympathetic nervous system, and renal sodium retention [11]. Excess growth hormone and insulin-like growth factor 1 that characterize patients with acromegaly have been shown to be associated with enhanced renal and extrarenal epithelial sodium channel activity [12].
Monogenic forms of hypertension are more frequent among children with hypertension. As shown in Table 31.3, these forms of hypertension are due primarily to specific mutations in the kidney affecting sodium or potassium transport systems and leading most of the time to increased sodium reabsorption as the main cause of the elevation of BP [13]. In accordance with this mechanism, patients with monogenic hypertension have a good BP response to diuretics.
Table 31.3
Most common forms of monogenic hypertension
Syndrome | K | pH | Renin | Aldo | Transmission | Gene | Treatment |
|---|---|---|---|---|---|---|---|
GRA | Low/N | High | Low | High | Autosomal dominant | Chimeric gene CYP11B1/11B2 | Spironolactone, amiloride, glucocorticoids |
Liddle’s syndrome | Low/N | High | Low | Low | Autosomal dominant | β and γ subunit of ENaC | Amiloride, triamterene |
AME | Low/N | High | Low | Low | Autosomal recessive | 11β-HSD | Spironolactone |
MR in pregnancy | Low | High | Low | Low | Unknown | Mutations of mineralocorticoid receptor | Delivery |
Gordon’s syndrome | High | Low | Low | Low | Autosomal dominant | WNK 1 and WKN4 | Hydrochlorothiazide |
HBS | N | N | N | N | Unknown | None |
In older children (>10 years), secondary forms of hypertension tend to become less frequent although their incidence remains higher than in adults. However, essential hypertension becomes more prominent in adolescents and its pathophysiology is probably closer, if not similar, to that of young and middle-aged adults as discussed below.
As discussed before, birth weight appears to be an important determinant of BP and hypertension in children and adolescents as suggested in the Barker’s hypothesis [4]. According to this hypothesis, the link between birth weight and hypertension is a low number of glomeruli and an incomplete renal maturity [5]. However, other factors related to the low birth weight may contribute to BP elevation including an immaturity of the corticoid system, a glomerular hyperfiltration, and a renal tubular adaptation leading to an overexpression of some renal transporters.
Taken together, it is suggested that hypertension in childhood and adolescence has a specific pathophysiology. The mechanisms of hypertension in this age category are important to consider because increasing evidence suggests that hypertension in infants and adolescents is associated with early target organ damage [14] and persistence of hypertension in adulthood.
31.3 Pathophysiology of Essential Hypertension in Adults
BP is the main driving force that brings blood to all organs of the body; thus, the control of BP is critical for life. In this context, it is not surprising that the regulation of BP is a highly complex and sometimes redundant interplay between renal, neural, cardiac, vascular, and endocrine factors modulated by genetic and environmental factors.
31.3.1 Role of Kidneys in the Pathogenesis of Essential Hypertension
Regulating sodium and water excretion and hence extracellular volume homeostasis, the kidney plays a crucial role in BP control. Indeed, as proposed by Guyton (1990), BP and sodium homeostasis are closely related through the pressure-natriuresis mechanism which enables to stabilize BP around a set point [15]. If renal perfusion pressure increases, renal sodium output increases and extracellular fluid volume contracts so that BP returns to its baseline value. According to this hypothesis, hypertension results from a shift of the BP set point due to a defect of the pressure-natriuresis mechanism. The regulation of sodium excretion in the pressure-natriuresis phenomenon occurs essentially in the proximal segments of the nephron and changes in medullary blood flow are important for the pressure-natriuresis relationship. In addition, many endocrine factors such as the renin-angiotensin-aldosterone system, nitric oxide, and prostaglandins can shift the pressure-natriuresis to higher or lower set points. There is also a good evidence that sodium handling by the distal segments of the nephron is critical to the regulation of sodium balance in relation to changes in BP and that other factors such as the sympathetic nervous system, a local intrarenal inflammation, and the generation of reactive oxygen species (ROS) can modify the pressure-natriuresis relationship [16].
The contribution of the kidneys to the development of hypertension has been demonstrated nicely using cross-transplantation studies in animals. These studies have demonstrated that transplantation of a kidney of a hypertensive rat into a normotensive animal leads to the development of hypertension in this latter. Conversely, transplantation of a normotensive kidney to a hypertensive rat normalizes BP showing BP follows the kidney [17]. More recent studies have used this technique in mice to investigate the role of the renin-angiotensin system in mediating the hypertension induced by cross-transplantation. These studies have demonstrated that AT1 receptors in the kidney but also in the vasculature contribute equally to the development of hypertension [18]. Some results have also been gathered in human transplantation, but the evidence obtained in these studies is less convincing as more confounding factors could interfere with the interpretation of the results [19].
In support of the role of kidneys in the pathophysiology of hypertension, one should mention once again the description of monogenic forms of hypertension which are associated to mutations in renal transport systems influencing renal sodium reabsorption in various segments of the nephron [20]. In essential hypertension, polymorphisms of some of these transport systems such as the epithelial sodium channel, Nedd4, or alpha-adducin may actually contribute to generate hypertension [21] or eventually protect against hypertension [22], but the frequency at which this occurs remains unknown.
31.3.2 Sympathetic Nervous System and Renin-Angiotensin-Aldosterone System
There are also several neuroendocrine systems that increase BP and maintain the hypertension state in part through their effects on renal function. The two most important are undoubtedly the renin-angiotensin-aldosterone and the sympathetic nervous systems. As both systems are discussed in separate chapters (Chap. 35, Sympathetic and Renin-Angiotensin Activity in the Pathophysiology of Hypertension; Chap. 36, Drugs Targeting RAAS in the Treatment of Hypertension and Other Cardiovascular Disease) of the present book, we shall only briefly summarize some effects of these systems as potential causes of hypertension. The renin-angiotensin-aldosterone system (RAAS), which originates with the synthesis of renin from the juxtaglomerular cells of the kidney, participates in the pathophysiology of hypertension in multiple ways through its vascular, endocrine, central, and renal properties. In recent years, it became apparent that the RAAS exists not only as a circulatory system but also as a local tissue system. As reviewed by Kobori et al. [23], many components of the renin-angiotensin-aldosterone system have been localized within the kidney. Thus, in addition to being a potent vasoconstrictor and a growth-promoting peptide, renal angiotensin II controls BP through its effect on renal hemodynamic and glomerular filtration rate as angiotensin II modulates the tone of both afferent and efferent arterioles. Angiotensin II also modulates renal tubular sodium reabsorption at different sites along the tubule through direct and aldosterone-mediated effects on sodium transport. But angiotensin II also induces inflammation, cell growth, mitogenesis, apoptosis, migration, and differentiation, regulates the gene expression of bioactive substances, and activates multiple intracellular signaling pathways, all of which may contribute to maintain a high BP and to promote the development of hypertension-induced complications. Interestingly, even a short-time exposure to angiotensin II may produce a long-term hypertension by provoking renal vascular lesions and a local inflammation [24, 25]. Of note, although an increased activity of renin-angiotensin-aldosterone system has been observed in many animal models of hypertension, not all hypertensive patients have an activated renin-angiotensin-aldosterone system; therefore, when measuring the activity of this system in hypertension, it should always be examined in the light of sodium intake as there is a strong interaction between sodium intake and the activity of the renin-angiotensin-aldosterone system.
Regarding the sympathetic nervous system, there is also abundant evidence that this system contributes to the regulation of BP and to the pathophysiology of hypertension [26]. Indeed, several animal models of hypertension are characterized by an increased sympathetic activity [27]. Moreover, in early stages of clinical hypertension, a combination of sympathetic hyperactivity and parasympathetic dysfunction has been observed in young subjects with an increased heart rate [28]. In the kidney, the sympathetic system has a major influence on renin secretion, renal hemodynamics, and tubular sodium handling [26]. The role of the sympathetic nervous system and of baroreflex control in the genesis of hypertension has led to the development of new interventional strategies to treat resistant hypertension (renal denervation, baroreflex sensitization) [29].
31.3.3 Prostaglandins
Another set of substances that regulate BP and may be involved in the genesis of hypertension are prostaglandins. Prostaglandins are the product of arachidonic acid metabolism [30]. Their production involves several steps: firstly, the release of arachidonic acid from membrane phospholipids under the action of phospholipase A2; secondly, the catalysis of arachidonic acid by cyclooxygenases (COX 1, 2, or 3) to generate PGH2; and thirdly, the generation of specific prostaglandins under the effect of prostaglandin synthases such as prostacyclin synthase leading to the formation of prostacyclin (PGI2) or thromboxane synthase which generates thromboxane A2 (Fig. 31.1). This cascade leads to the synthesis of several prostaglandins with multiple biological properties, including vascular, renal, and inflammatory effects. Phospholipase A2 is activated by a variety of stimuli, including angiotensin II, norepinephrine, and bradykinin.
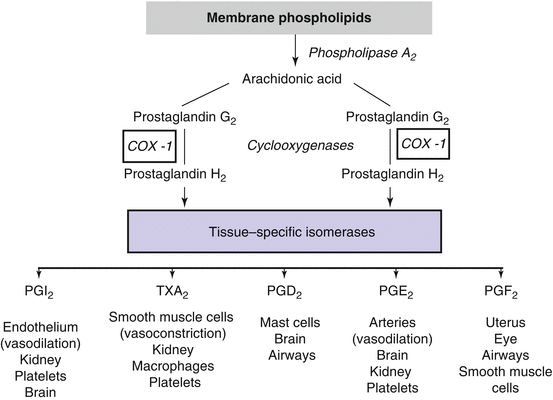
Fig. 31.1
Metabolism and systemic effects of prostaglandins. Schematic representation of the metabolism of prostaglandins and their effects on various tissues. Note that some prostaglandins can affect vascular tone and hence blood pressure through vasodilating prostaglandins (PGI2 and PGE2) as well as through the vasoconstrictor effect of thromboxane A2 (TXA2)
Prostaglandins act mainly near their sites of release because they are degraded rapidly by local metabolism into inactive products. At the vascular level, prostaglandins can produce either a vasoconstriction (thromboxane A2, PGF2α) or a vasodilatation (PGE2, prostacyclin). One important property of vasodilatory prostaglandins is their ability to modulate the vasoconstriction induced by potent vasoactive substances such as angiotensin II. In the normal adult kidney, both COX-1 and COX-2 are constitutively expressed. COX-1 is expressed in the glomerulus, the afferent arteriole, and tubular cells, and COX-2 has been expressed in the afferent and efferent arterioles, the podocytes, the macula densa, and some tubular and interstitial cells. Intrarenal prostaglandins participate actively in the regulation of renal perfusion and glomerular filtration rate. They are also implicated in the maintenance of sodium, potassium, and chloride homeostasis and in the regulation of renin secretion [31, 32]. The impact of prostaglandins on vessels, renal electrolyte balance, and renin secretion may be of particular relevance to the genesis of hypertension [33]. Indeed, data have demonstrated that mice deficient in the PGI2 receptor are resistant to the development of renovascular hypertension, a renin-dependent form of hypertension [34].
In hypertensive patients, a deficiency in vasodilatory prostaglandins has been reported [35], and an increase in thromboxane A2 has been found in essential hypertension [36]. These observations lead to the hypothesis that there might be an imbalance between anti- and pro-hypertensive prostaglandins in hypertension. The potential role of prostaglandins in hypertension is further emphasized by the observation that selective and nonselective COX-1 and COX-2 inhibitors increase BP, favor sodium retention, and may cause hypertension in some patients [37]. However, it appears that the hypertensive effect of nonsteroidal anti-inflammatory drugs is more frequent among hypertensive than normotensive patients. This would indicate that prostaglandins act as a counter-regulatory mechanism to limit the increase in BP in hypertension and may not be a primary hypertensive mechanism [35].
31.3.4 Role of Vasculature in the Pathogenesis of Essential Hypertension
As observed by Folkow in the 1970s [38], when BP increases persistently, blood vessels undergo an adaptation characterized by a structural remodeling leading to an increase in wall:lumen ratio. This adaptation actually maintains a chronic increase in vascular resistance in established hypertension. Clinically, it appears that this vascular remodeling occurs very early in the course of the pathogenesis of hypertension probably even before BP has reached the hypertensive range [39]. Today, several investigators have actually confirmed using gluteal biopsies that there is indeed a remodeling of small arteries in hypertensive patients, that small vessel remodeling may have a major impact on target organs, and that antihypertensive therapy can reverse it [40]. The mechanisms involved in this remodeling imply essentially an increased vascular tone and an increased activity of the sympathetic nervous system, angiotensin II which causes a proliferation of smooth muscle cells, endothelin-1, and perhaps inflammatory processes [41]. At the cellular level, the matrix rearrangement appears to involve integrins and tissue transglutaminase, one of a family of transglutaminase enzymes, which includes factor XIII [42].
31.3.4.1 Nitric Oxide
The endothelial layer of the vascular wall produces several substances, which are closely involved in the regulation of the circulation and vascular wall homeostasis. Some of these substances are direct vasodilators such as nitric oxide, prostacyclin, and endothelial-derived hyperpolarizing factor, and some are potent vasoconstrictors such as endothelin-1 and thromboxane. Nitric oxide is formed by the enzyme nitric oxide synthase (NOS) from the amino acid L-arginine. Once formed, NO diffuses to the underlying vascular smooth muscle cells, activates soluble guanylyl cyclase, and produces vasorelaxation [43]. Three forms of NOS have been described: a neuronal NOS present in neural cells, an inducible NOS (iNOS), and an endothelial NOS (eNOS) present essentially in the endothelium. In the vessels, NO is released from endothelial cells in response to physical stimuli (shear stress and hypoxia) and by the stimulation of endothelial receptors such as bradykinin and muscarinic receptors. NOS activity can be inhibited using endogenous analogues of L-arginine such as asymmetric dimethylarginine or N-monomethyl-L-arginine (L-NMMA). Some of these analogues are increased in disease states, for example, in patients with chronic renal failure.
The potential role of NO in the pathophysiology of cardiovascular diseases including hypertension has first been evoked with the demonstration that acetylcholine, in the absence of endothelium, is a vasoconstrictor rather than a vasodilator [44]. Following this seminal observation, numerous studies have demonstrated the crucial role of the endothelium in the regulation of cardiovascular homeostasis and have led to the concept of endothelial dysfunction. Thus, endothelial dysfunction of large and small arteries has been described in animal models of hypertension as well as in patients with essential hypertension [45–47]. Several mechanisms are discussed whereby endothelial dysfunction results in hypertension. Studies have suggested an impaired NO synthesis and release, but endothelial dysfunction may also be consecutive to an increased breakdown of NO as the vasorelaxing properties of NO are counteracted by oxidative processes in tissues. Superoxide anions are potent scavengers of endothelial-derived NO. Whether endothelial dysfunction is a primary or secondary event in hypertension has been discussed because an elevation of blood pressure per se can cause endothelial dysfunction [48]. However, a blunted endothelium-dependent vasodilatation has been observed in offspring of hypertensive parents suggesting that endothelial dysfunction can precede the development of hypertension and may play a primary role in the genesis of the disease [49]. Of note, transgenic mice deficient for the eNOS develop a systemic hypertension associated with an increased peripheral vascular resistance [50, 51]
The role of nitric oxide in the pathogenesis of hypertension is controversial [52]. Nitric oxide can contribute to the etiology of hypertension by various other mechanisms, one of them being the development of atherosclerosis. Indeed, NO has been reported to decrease monocyte and leukocyte adhesion to endothelial cells, to inhibit platelet aggregation and platelet-vessel wall interaction, to decrease the transport of lipoproteins into the vessel wall, and to inhibit vascular smooth muscle cell proliferation as well as some components of the vascular inflammation. Another pathway by which NO can affect blood pressure is the interaction with the renin-angiotensin system. Nitric oxide has indeed been found to suppress renin release by juxtaglomerular cells and hence to reduce the activity of the renin-angiotensin system and to participate in the regulation of renal hemodynamics [53]. As discussed previously, NO also interacts closely at the vascular level with endothelin to limit the vasoconstrictor effect of endothelin. Some of these interactions may actually lead to an increased vascular tone. At last, the activity of nitric oxide is related to other vasoactive compounds such as prostanoids and bradykinins.
31.3.4.2 Endothelin
Endothelin (ET) is a very potent endogenous vasoconstrictor produced by the endothelium and identified by Yanagisawa et al. in 1988 [54]. There are three isoforms of endothelin (i.e., ET-1, ET-2, and ET-3), but endothelin-1 is the only relevant peptide in humans. Endothelin is derived from pro-endothelin, which is cleaved into a big endothelin and then converted to the active endothelin-1 by an endothelin-converting enzyme. Several stimuli induce endothelin release by endothelial cells including shear stress, thrombin, angiotensin II, vasopressin, catecholamines, and hypoxia [55]. The effects of endothelin are mediated by two receptors, i.e., the ET-A and ET-B receptors. The ET-A receptor is widely distributed and it is the principal receptor located on vascular smooth muscle cells and cardiomyocytes. In these cells, activation of ET-A receptors leads to an activation of phospholipase C, to an increase in intracellular calcium, and, hence, to cell contraction. The ET-B receptor is located on both vascular smooth muscle and endothelial cells. In endothelium cells, activation of ET-B receptors releases vasodilating substances such as nitric oxide (NO), prostacyclin, and adrenomedullin. In muscle cells, activation of the ET-B receptor induces a vasoconstriction (Chaps. 34 and 45).
If vasoconstriction is the hallmark of endothelin’s action, several other biological properties of endothelin have been described. Thus, renal function appears to be particularly responsive to the effects of endothelin [56]. Administration of low doses of endothelin-1 in animals and humans has been shown to decrease glomerular filtration rate and renal blood flow through the stimulation of vascular smooth muscle cells and contraction of mesangial cells and to reduce urinary sodium excretion. Similarly, overexpression of endothelin in the mouse kidney has been associated with the development of glomerulosclerosis, the development of interstitial fibrosis, and the development of renal cysts but not hypertension suggesting a role of endothelin in the development of some renal diseases independent of the hypertensive effect. These effects appear to be mediated by the activation of ET-A receptors. However, endothelin-1 can also lower BP and produce a natriuretic response through the activation of ET-B receptors which have been localized on renal tubular epithelial cells [57]. Recent data suggest that the renal medullary endothelin system is important for BP regulation. Indeed, transgenic rats deficient in endothelin-1 specifically in the collecting duct develop hypertension [58]. In the normal guinea pig heart and in isolated cardiomyocytes, endothelin-1 had positive inotropic and growth-promoting effects [59]. At the vascular level, as mentioned earlier, endothelin may contribute to the remodeling of small and large arteries [60].
Experimental data have demonstrated that endothelin-1 interacts very closely with nitric oxide via activation of ET-B receptors on endothelial cells. Indeed, endothelin-1 promotes the release of NO and thereby maintains a balance between the vasodilatory effect of NO and the vasoconstrictor effect of endothelin-1 itself. There is also a close interaction between endothelin and the renin-angiotensin-aldosterone system [61]. Angiotensin II enhances the vascular responsiveness to exogenous endothelin-1 and increases the release of endothelin-1 and the expression of preproendothelin in endothelial cells.
The role of endothelin as a potential pathogenic factor in hypertension has been suggested using several experimental and clinical approaches [60]. In rats, knockout of the ET-B receptor is associated with the development of severe salt-sensitive hypertension [62]. In humans with essential hypertension, plasma endothelin levels are usually not elevated except in Afro-Americans [63]. However, circulating concentrations of endothelin may not necessarily reflect the tissue concentrations because endothelin acts as a paracrine/autocrine system. Nonetheless, elevated circulating concentrations of endothelin have been reported in some human and experimental forms of hypertension such as the mineralocorticoid-induced and renovascular hypertension in the rat and hypertension in renal transplant patients and patients with diabetes or chronic renal failure. There are also data suggesting that endothelin plays a role in the development of hypertension in pregnancy-induced hypertension [64, 65].
The best demonstration of the role of endothelin in hypertension in humans has come from the use of selective endothelin antagonists. Indeed, in recent years, several selective and nonselective non-peptide antagonists of ET-A and ET-B receptors have been developed and investigated. In mild to moderate hypertensive patients, the dual ET-A and ET-B receptor antagonist bosentan (500–2,000 mg) lowered BP as effectively as the angiotensin-converting enzyme inhibitor enalapril (20 mg) [66]. Similarly, a significant decrease in blood pressure was found with darusentan, a selective ET-A endothelin receptor antagonist, in patients with a mild to moderate hypertension [67].
31.3.5 Metabolic Factors in the Pathogenesis of Essential Hypertension
31.3.5.1 Obesity
Essential hypertension is a common feature in patients with obesity, metabolic syndrome, and diabetes. Indeed, in obese subjects, hypertension is diagnosed in up to 50 % of patients, and similarly about 50 % of type 2 diabetic patients are hypertensive at the time of diagnosis [68, 69]. The pathogenesis of hypertension in these metabolic disorders has some specificity and implies, in addition to the sympathetic and renin-angiotensin systems, other mechanisms such as hyperglycemia, insulin resistance, adipokines (leptin and adiponectin) play.
In obese patients with hypertension, the elevated BP is associated with sodium retention and rightward shift of the pressure-natriuresis relationship. This shift is due in part to a sustained activation of the renin-angiotensin system and an increased sympathetic activity but also to the physical compression of the kidneys by surrounding visceral fat and increased renal sinus fat [70]. Considering the role of the renin-angiotensin system in obesity-induced hypertension, aldosterone appears to be particularly important to mediate an increase in BP observed in obesity as well as in patients with metabolic syndrome. Indeed, higher plasma aldosterone levels have been measured in obese patients and patients with the metabolic syndrome [71]. In the study by Goodfriend et al. (2004), oxidative stress and oxidized fatty acids derived from linoleic acid stimulated aldosteronogenesis and thereby promoted sodium retention [72]. These oxidized fatty acids are essentially produced by visceral fat, an observation that confirms that visceral fat contains all the components of the renin-angiotensin system.
31.3.5.2 Leptin
Regarding the sympathetic nervous system in obesity, many factors contribute to increase its activity including angiotensin II, baroreflex sensitivity, hyperinsulinemia, and sleep apnea but also hypoghrelinemia and hypoadiponectinemia and leptin resistance. An interesting observation made in animal models of obesity is that sympathetic tone is only moderately increased and that not all organs demonstrate an elevated sympathetic activity [73]. Thus, some studies have shown that sympathetic drive is increased in the kidney and muscle but not necessarily in the heart [73]. Moreover, visceral fat rather than subcutaneous fat appears to participate in the increased sympathetic nerve activity. Leptin and adiponectin, the two molecules produced by adipocytes, may actually contribute to an increase in BP observed in obese patients through their effects on the sympathetic and renin-angiotensin systems. Plasma levels of leptin are proportional to the fat mass. Leptin acts on a specific leptin receptor localized in the brain where its stimulation reduces appetite and increase energy expenditure, thus lowering body weight [74]. Mice deficient in leptin develop hyperphagia, obesity, and insulin resistance. Leptin has also pro-inflammatory properties, which may contribute to the high cardiovascular risk associated with obesity. The ability of leptin to increase BP has been suggested by studies in which leptin was infused either in the brain or peripherally. These leptin administrations actually activated the sympathetic nervous system and progressively increased BP, an effect that could be blocked by administration of an alpha- and beta-adrenergic blocker [75]. In obese humans and rodents, elevated levels of leptin have been measured but surprisingly anorexia is generally absent. Therefore, it has been suggested that leptin resistance explains this discrepancy and that this resistance is frequent in obesity. Thereafter, the observation in animals of dissociation between the ability of leptin to stimulate sympathetic tone and to increase BP and the finding of an absence of simultaneous reduction of appetite and body weight has generated the concept of selective leptin resistance [76] as shown in Fig. 31.2. The concept of leptin resistance has been reviewed recently and the mechanisms of this dissociation are discussed in details by Mark et al. [77]. Of note, the actual role of leptin in mediating the obesity-induced hypertension in humans remains highly controversial as many conflicting results have been published [77].
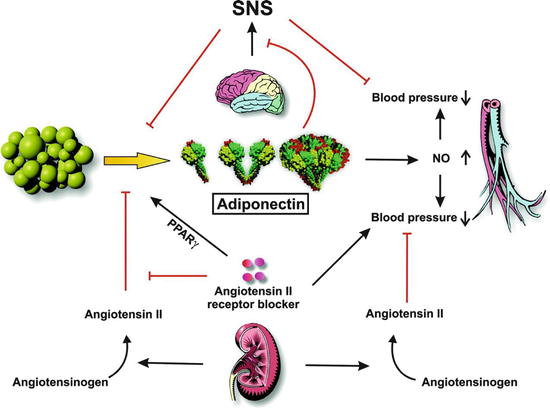
Fig. 31.2
Role of adiponectin in blood pressure control (Adapted with permission from Ref. [78]). Adipose tissue secretes three molecular forms of adiponectin into circulation. Adiponectin stimulates the production of NO, which is involved in the regulation of blood pressure. Renin secreted from the kidney cleaves angiotensinogen to produce angiotensin II. Angiotensin II plays an inhibitory role in adiponectin production. Angiotensin II AT1 receptor blockers display pleiotropic effects on blood pressure control through receptor blocking and adiponectin stimulation. SNS overdrive increases blood pressure and inhibits the production of adiponectin. Also, adiponectin inhibits the activation of SNS through central actions. Black arrows indicate activation and red lines indicate inhibition
31.3.5.3 Adiponectin
Adiponectin is another complex molecule secreted by adipocytes which has been associated with metabolic and cardiovascular diseases including hypertension [78] (Fig. 31.2). This molecule is present in the plasma in three molecular forms (high-molecular-weight, low-molecular-weight, and trimeric forms). Adiponectin acts on two distinct receptors localized in various tissues (muscle, liver, endothelium) where it can mediate the effects on peroxisome proliferator-activated receptors (PPAR), AMP kinase activation glucose uptake, and beta-oxidation [78]. In endothelial cells, adiponectin has also been shown to increase NO production by regulating endothelial NOS activity [79]. In obesity, low levels of adiponectin have been measured suggesting that hypoadiponectinemia is associated with an increase in BP. In animals, there is a close relationship between angiotensin II and the sympathetic nervous system and adiponectin. In rats, angiotensin II infusion decreases plasma adiponectin levels and inhibition of the renin-angiotensin system increases adiponectin through the PPARγ nuclear receptor [78]. As noted earlier, adiponectin is closely related to the sympathetic system. An increased sympathetic tone is associated with a reduction of plasma adiponectin and this may influence BP. The interaction between these two systems occurs in the periphery as well as in the brain and may be involved in the genesis of hypertension in obesity [80].
31.3.5.4 Insulin
Insulin is another hormone that is increasingly considered in the genesis of hypertension in obese patients and patients with the metabolic syndrome [81]. Hypertension, obesity, dyslipidemia, and glucose intolerance represent a cluster of risk factors which is called metabolic syndrome although this entity is often criticized. Insulin resistance in some peripheral tissues appears to be the main feature of metabolic syndrome. The peripheral insulin resistance is associated with an increased activity of the sympathetic nervous system and endothelin and a decrease in NO production [82]. Moreover, in the kidney, insulin has been shown to cause sodium retention, an effect which may further contribute to increased BP in this clinical context.
31.3.6 Other Hormonal Factors in the Pathophysiology of Essential Hypertension
In addition to the many system described above, several other hormonal factors may participate in the regulation of BP either through their vasodilating properties, which may be altered in hypertension, or through their vasoconstrictor effects. In this respect, one should consider natriuretic peptides, kinins, vasopressin, and dopamine.
31.3.6.1 Natriuretic Peptides
Natriuretic peptides were identified in the 1980s with the observation that rat atrial extracts had potent natriuretic and vasodilatory properties [83]. This original finding led to the identification of the atrial natriuretic peptide (ANP) and subsequently to the recognition of a family of four distinct natriuretic peptides: ANP (17 amino acids), BNP (32 amino acids), CNP (22 amino acids), and urodilatin (32 amino acids). ANP is synthesized and secreted predominantly by the atria. BNP was initially isolated from pig’s and dog’s brains but it is produced essentially by cardiomyocytes [84]. CNP has been localized in the brain and in the heart but also in several other peripheral tissues including the kidney, the adrenal glands, and the endothelium. ANP and BNP are released from the heart in response to changes in atrial or ventricular stretch. Plasma levels of natriuretic peptides are also influenced by the body position and the salt intake. Natriuretic peptides act by stimulating specific receptors (natriuretic peptide receptors A, B, and C). These receptors are widely distributed throughout the body including endothelium, smooth muscle cells, heart, adrenal gland, lung, brain, adipose tissue, and kidney [85]. Natriuretic peptides are degraded by the neutral endopeptidase [11, 24] and by a receptor-mediated clearance via the C receptor. Inhibition of neutral endopeptidase to increase plasma natriuretic peptides is today one approach to treat hypertension and heart failure in combination with a blocker of the renin-angiotensin system [86].
ANP and BNP possess diuretic, natriuretic, vasodilatory, and anti-hypertrophic, antifibrotic, antiproliferative, and anti-inflammatory properties. ANP also causes intravascular volume contraction as documented by increases in hematocrit and serum albumin when administered to binephrectomized rats [87]. ANP has an inhibitory action on aldosterone and renin secretion [88]. ANP and BNP antagonize vasoconstriction induced by norepinephrine or angiotensin II. There is also some evidence that the central effects of ANP contribute to fluid and electrolyte balance and to the regulation of systemic hemodynamics [89]. These central effects of ANP are mediated by an interaction between ANP and sympathetic tone in the brain stem.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


