Fig. 3.1
Schematic depiction of the interrelated and (usually) opposing neurohormonal mechanisms/systems activated in acute and in chronic HF and their (patho)physiological actions for the circulation. LV left ventricular, CO cardiac output, BP blood pressure, AVP arginine vasopressin, MMP matrix metalloproteinase, TNFα tumor necrosis factor-alpha, NE norepinephrine, Epi epinephrine, DA dopamine, Ang angiotensin, HR heart rate, CRF corticotropin-releasing factor. See text for details
3.2 MI-Coronary Atherosclerosis and HF
Atherosclerosis is the gradual accumulation of lipids, blood cells, calcium, and other materials on the walls of the arterial vasculature. Stable plaque buildup can eventually cause ischemia, while unstable plaques, which are prone to rupture, can lead to thrombus formation or can detach, travel, and occlude downstream arteries [1]. Therefore, atherosclerosis has acute and long-term consequences on the body’s vasculature ranging from acute coronary syndrome, sudden cardiac death, arrhythmias, transient ischemic attacks to cerebrovascular disease and beyond [2] (Chap. 25).
In the heart, one of the most serious manifestations of atherosclerotic plaques is that of the acute coronary syndrome known as ST-elevated MI (STEMI) or simply MI [3]. MIs are caused by complete occlusion of the arteries supplying the heart directly leading to myocardial tissue damage. In fact, the most common cause of MIs is ischemic heart disease, and in turn, MIs are one of the leading causes of HF because they directly alter the structure and function of the heart [4, 5]. Due to major advances in medicine, mortality due to MIs has decreased while complications such as recurrent MIs, arrhythmias, atrial fibrillation, and HF are on the rise [1, 6]. The incidence of post-MI HF has been reported to range from 5 to 50 % depending on the population being studied and study design [7–9]. However, up to half of patients may already have some degree of transient or chronic HF or may develop it while hospitalized for the MI. Others will likely develop it within 30 days of hospital discharge. Moreover, 40 % of MI patients will develop left ventricular systolic dysfunction after discharge. The progression to HF following an MI is more likely to occur in older patients, females, those with an anteriorly located infarct, and in individuals with coexisting comorbidities such as hypertension and diabetes (Chaps. 19 and 20).
HF often occurs as the terminal common pathway of any combination of cardiac conditions that affect the myocardium’s productivity. It can be described as the inability of the heart to adequately fill or contract in order to meet the body’s metabolic demands. MIs affect the ability of the heart to properly fill and eject blood depending on the location and extent of infarct. Directly following an MI, persistent ischemia directly leads to cardiomyocyte loss from necrosis, apoptosis, inflammation, and neurohormonal mechanisms which all contribute to cardiac remodeling as well as with long-term detrimental consequences (Fig. 3.1) [10–19]. Even with successful and timely revascularization after an MI, the occurrence of microvascular injury remains high. Critical factors for injury and repair processes are age, time to treatment, exaggerated platelet activation, presence of diabetes or other comorbidities, and severity of atherosclerotic heart disease [20, 21]. Induced by ischemia, cardiomyocyte death by apoptosis and necrosis peaks within 8 h after an infarction. While necrosis may be more prominent in the acute phase of MI, apoptosis seems to play a more important role in the long run.
Inflammatory processes play an important role in the body’s response to an MI and are especially active in the first 2 weeks following the event. Ischemic-reperfusion injury or microvascular damage following restoration of blood flow, oxygen, and nutrients from the generation of free radicals, leukocyte, and inflammatory response to the infarcted tissues is a well-established phenomenon. Macrophages and neutrophils are rapidly recruited by chemokines and cytokines such as interleukins 1b, 6, 8, and 18 and tumor necrosis factor-α (TNFα) in order to initiate the healing process by phagocytosing necrotic cells, producing matrix metalloproteinase (MMP) to break down the extracellular matrix (ECM), secreting myeloperoxidase and other compounds [22–24]. Reactive oxygen species such as superoxide and peroxynitrite generation from inflammatory cells increases while there is decreased antioxidant activity furthering cardiomyocyte death [22]. Other mediators of inflammatory cells such as the pro-fibrotic cytokines tissue growth factor-beta1 (TGF-β1) and connective tissue growth factor (CTGF) are also involved in the clearing of necrotic cells [25–32]. However, an overwhelming inflammatory and immune response can also have deleterious effects on the affected myocardium such as by the generation of toxic by-products blocking sufficient perfusion of the infarcted areas [33, 34].
The RAAS, the SNS, and the vasopressin systems along with their effector hormones and compounds also play a pivotal role in the development of HF [2, 16] (Chaps. 5 and 35). As the main product of RAAS, angiotensin II is involved in regulating electrolytes and fluid volume, both of which have important consequences for the signs and symptoms of HF. Local angiotensin II and aldosterone concentrations are also increased in an infarcted heart influencing fibrosis and hypertrophy [2]. Aldosterone has been particularly associated with coronary vasculature damage as well as left ventricular dysfunction and promotion of fibrosis [2]. Higher levels of the catecholamines epinephrine and norepinephrine following MI are known to damage the endothelium and are associated with subsequent development of HF as well as death [21]. Furthermore, circulating levels of particular compounds may be important prognostic markers of progression to myocardial damage, development of complications, and mortality. Peak C-reactive protein (CRP) levels may also predict negative outcomes from an MI including HF [10]. Finally, serum levels of soluble tumor necrosis factor receptor (sTNFR), which is an important mediator of inflammation and apoptosis, have been found in post-MI patients to correlate well with the size of infarct and extent of left ventricular dysfunction [22].
Ventricular remodeling is strongly related to cell death, to inflammation, and to the neurohormonal responses of an MI (Fig. 3.2) [34]. It begins within days of an MI and may continue for months after the initial insult to the myocardium. Inflammatory and immune cells such as macrophages and neutrophils together with their pro-fibrotic cytokines can cause changes in the extracellular matrix due to collagen synthesis and deposition through myofibroblasts [34]. Because myocardial tissue has inadequate ability to regenerate, structural consequences of an MI often include hypertrophy of viable tissue due to collagen remodeling as well as myocardial scarring and stiffness in infarcted areas [34]. Ventricular remodeling therefore primarily consists of thinning at the infarcted sites, hypertrophy of the viable myocardium, and ventricular dilation. Fibrosis proximal and distal to the infarcted sites leads to scar formation and wall stiffness and is influenced by the presence of aldosterone and angiotensin II in addition to aforementioned compounds [2]. Both collagen generation and degradation in the extracellular matrix occur following an MI which will inevitably lead to changes in the shape and capacity of the affected heart chamber [2, 34].
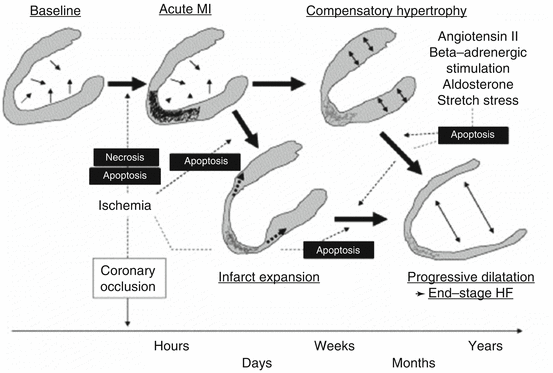
Fig. 3.2
Time course of ventricular remodeling and progression to HF (Reproduced with permission from Ref. [6])
In summary, myocardial injury can lead to cell death, inflammation, and neurohormonal responses, triggering pathological cardiac remodeling with deleterious long-term consequences such as HF. These adaptive mechanisms are an effort by the injured heart to maintain adequate cardiac output sufficient to adequately perfuse the body. However, their continued activation contributes to worsening cardiac function as the heart progressively fails in its ability to maintain cardiovascular homeostasis and the patient progresses through the stages of HF.
3.3 Hypertension and HF Pathophysiology
Elevated blood pressure affects more than 65 million adult Americans and is a major risk factor for MI, stroke, renal failure, and HF [35, 36]. Long-term management of both systolic and diastolic hypertension can reduce the risk of developing HF by approximately 50 % [37–39]. Because HF due to hypertension is preventable, it is of paramount importance to educate those at risk on antihypertensive medications and lifestyle modifications. Activities such as weight loss, control of sodium intake, and exercise are steps in the right direction which reduce or eliminate progression to HF [40]. Before discussing the mechanisms by which chronic hypertension may lead to HF, it is essential to review the diagnostic criteria of hypertension and the particular categories of this chronic illness.
The recently released JNC8 guidelines define hypertension by age class. Those younger than 59 years old are considered hypertensive if presented with either a systolic blood pressure greater than 140 mmHg or a diastolic blood pressure above 90 mmHg. Those greater than 59 years old require a systolic pressure above 150 mmHg or a diastolic pressure greater than 90 mmHg to be diagnosed as hypertensive [41]. The cause of heightened blood pressure may also be used as a means to define the condition. A diagnosis of primary hypertension is given when the etiology is of unknown origin and is present in approximately 90–95 % of diagnosed individuals; whereas, a diagnosis of secondary hypertension implies that the origin is known and is found in 5–10 % of those diagnosed [42]. Although there are differences between primary and secondary hypertension, the key pathophysiologies behind the two are similar and will be discussed as one (Chap. 30). This section will focus on hypertension-induced mechanisms leading to the eventual development of HF.
Cardiac output is a common biological marker used to diagnose HF. An increased stroke volume, heart rate, and blood pressure or decreased peripheral vascular resistance leads to heightened cardiac output. An increase in cardiac output, brought about through increased blood pressure, puts a strain on cardiomyocytes and may ultimately lead to cell injury and loss of elasticity from collagen deposition. This accumulation of collagen is due to changes in the myosin heavy chain causing a reduced contractility [43]. Loss of the myocardial cell count present in chronic, untreated hypertension is central to the development of HF. Furthermore, the damage induced by hypertension to the cardiac muscle often leads to an inflammatory response which perpetuates cell damage and loss. The overall loss of cell contractility leads to decreased stroke volume, cardiac output, and ejection fraction of the heart [19]. Once the heart’s ejection fraction, or the amount of blood being pumped from the heart, falls to a level below 40 %, HF with reduced ejection fraction is diagnosed [44].
Certain populations pose with an increased risk of developing chronic HF as a result of hypertension. Age-related changes in the cardiovascular system place older adults in such a category [45]. Typically, a rise in blood pressure is sensed by the smooth muscles’ lining vessel, which releases nitric oxide leading to vessel dilation and reduced resistance. Elasticity in blood vessels is gradually lost due to aging making it increasingly difficult to adjust vessel diameter and account for changes in blood pressure [46]. These vessels are constantly constricted causing increased pressure that erodes the cardiomyocytes and increases fibroblasts, collagen, and hypertrophy of cardiac tissue. Age-related changes alter ATP concentration, resulting in higher levels of calcium ions which increase norepinephrine and further stress the heart (Chap. 4). Finally, the RAAS has the potential to alter blood pressure levels, especially in males and in African-American populations. Hence, there are many mechanisms by which hypertension may ultimately lead to HF [43].
3.4 Renal Dysfunction and HF
An important bidirectional relationship exists between the kidneys and the heart. Each organ functions to maintain a homeostatic balance allowing the other organ to maintain optimal efficiency. Conversely, dysfunction in either organ system has the ability to cause or propagate disease in the other organ by means of common feedback pathways [47]. The clinical significance in this relationship has been studied and shown through observations of higher percentage of patients with cardiovascular disease in patients with a chronic kidney condition [48–51]. The 2013 US Renal Data System Report shows that 41 % of patients with stage 4 or 5 chronic kidney disease (CKD) developed HF compared to 7 % in non CKD patients in Medicare patients age 66 years or older [52]. Impaired renal function is also associated with an increased risk of hospitalization due to HF as well as an increased risk of cardiovascular-related deaths [47, 53, 54].
Under normal conditions, the kidneys are perfused by the heart. In other words, the kidneys are reliant upon cardiac output for normal function. The kidneys sense the level of perfusion from the heart and adjust the blood pressure accordingly through the RAAS. In the presence of low perfusion or blood pressure (reduced cardiac output), the RAAS is activated to release renin from the juxtaglomerular apparatus of the kidneys. Renin cleaves angiotensinogen circulating from the liver to angiotensin I. Angiotensin-converting enzyme converts angiotensin I to the biologically active angiotensin II [55]. The effects of angiotensin II include direct vasoconstriction, sodium retention through a direct renal tubular effect and increased aldosterone secretion, and stimulation of water intake through the thirst center [56]. The net result of these physiologic mechanisms is an increase in fluid volume and blood pressure. Under normal physiological conditions, this mechanism between the heart and kidneys maintains blood pressure within a set range. It is when this system falls out of homeostatic balance that a dangerous cycle of failure in each system is observed (Chaps. 35 and 36).
In the presence of altered kidney function, the kidneys lose the ability to maintain sodium, potassium, water, and acid–base balance. This balance is lost due to improper filtering and clearance mechanisms within the kidney [57]. In relation to the cardiorenal system, these altered mechanisms lead to an imbalance of sodium. Water follows this increase of sodium resulting in an increase in intravascular volume. This increase in volume sequentially leads to a prolonged sense of hypoperfusion by the kidneys. Renal mechanisms compensate for this decreased perfusion by continuous activation of the RAAS. Once activated, the RAAS adds to the systemic imbalance of electrolytes, fluids, and intravascular volume by increasing aldosterone secretion. The activation of RAAS also leads to direct vasoconstriction, worsening the decline in the cardiorenal cycle. Increased levels of plasma renin and aldosterone are seen in patients with severe HF in whom the activity of the RAAS is increased [58].
The prolonged inability to stop this cycle of decreased renal perfusion and increased ventricular vascular resistance leads to the development and worsening of HF. The initial decrease in cardiac output leads to an inability of the kidneys to inhibit the RAAS. This system remains activated because of a constant sense of hypoperfusion by the kidneys. The prolonged activity of this system leads to several compounding factors affecting cardiac function including sodium and water retention, intravascular volume expansion, sympathetic activation, and electrolyte and acid–base imbalances (Fig. 3.3) [47, 60]. The body is fixed in a persistent hypertensive state coupled with increased intravascular volume, causing an increase in vascular resistance. This increase in vascular resistance increases cardiac workload. The initial mechanism of compensation in HF is activation of the SNS. In HF, peripheral alpha2-adrenoceptor function is lost contributing to higher levels of circulating norepinephrine [59]. Increased norepinephrine levels lead to an increase in cardiac inotropy and chronotropy as well as peripheral vasoconstriction. Though this compensatory mechanism is initially useful in maintaining stroke volume and cardiac output, eventually, the increased workload of the heart leads to negative effects on the heart muscle [56]. The additional stress placed on the heart can lead to myocyte hypertrophy and dysfunction [61]. Alterations of myocytes are fundamental in the cardiac remodeling process. When exposed to stress, the myocytes produce proteins altering their shape and structure in an attempt to adapt to maintain stroke volume and cardiac output. These alterations lead to increased ventricular wall thickness and further deterioration of cardiac function [62]. Research has led to the theory that angiotensin II may directly lead to myocyte hypertrophy, as well [63]. Progression of HF is dependent upon the severity and duration of these physiologic mechanisms. The predominant contribution of angiotensin II in HF development and progression illustrates the clinical importance of ACE inhibitors and angiotensin receptor blockers (ARBs) in the treatment of HF, wherein RAAS antagonists have shown to decrease mortality [64].
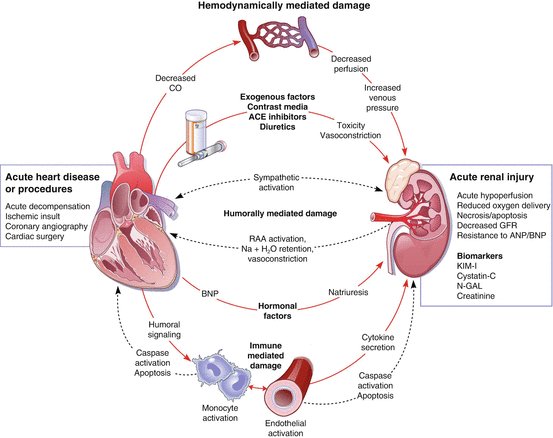
Fig. 3.3
Interplay between kidney injury (renal failure) and HF pathogenesis (Reproduced with permission from Ref. [59])
3.5 Central Nervous System (CNS) Disorders and HF
Approximately one in five HF patients is diagnosed with depression [65, 66]. Some studies have observed this number to be higher depending on the study and assessment method used [66, 67]. The incidence of a comorbid depressive condition increases with worsening stages of HF with as many as 42 % of patients observed with this comorbidity in patients with NYHA class IV HF [68]. This patient population has also been observed to have worse clinical outcomes in terms of greater hospitalization and rehospitalization rates as well as increased morbidity and mortality [66–69]. Furthermore, depression has been associated as an independent predictive factor for hospital readmissions and mortality in HF patients, as well as increased health-care-associated costs and diminished quality of life [70–73]. Anxiety disorders are not as prevalent appearing in roughly 15 % of patients depending on the study and assessment tools used, but this percentage is still higher than the general population percentage [67, 74]. Depression and anxiety can both contribute to the development and progression of HF, as patients with these comorbid conditions become fixed in this pathophysiologic loop of disease progression [75].
The effects of depression potentiate a cascade of events which both directly and indirectly contribute to worsening cardiac function and cardiovascular disease progression. The mechanisms by which depression and anxiety lead to the development and progression of HF are precipitated through the activation of the hypothalamic–pituitary–adrenal (HPA) axis, dysregulation of the autonomic nervous system, impaired platelet function, and activation of the cytokine cascade [67, 68, 73, 76]. Overstimulation of the HPA axis increases the level of cortisol in the circulation leading to hypertension, increased levels of serum lipids, and insulin resistance, which all play a role by either direct or secondary pathways in worsening of cardiovascular disease [77]. Overstimulation of the HPA axis also causes an increase in sympathetic activity. As discussed in other pathological mechanisms of HF, an increase in circulating catecholamines first leads to maintenance of cardiac output but eventually leads to prolonged vasoconstriction and hypertrophy of the cardiac cells [67]. Increased sympathetic activity and decreased parasympathetic activity lead to increased heart rate and decrease heart rate variability (HRV) [77]. Decreases in HRV predispose patients to arrhythmias thus increasing the mortality in HF patients [76]. The prolonged increase of sympathetic activity and circulating cortisol also leads to the development of a hyper-coagulation state via increases in clotting factors, factor III and von Willebrand factor, and increases in circulating norepinephrine [75, 77] (Chap. 23). Researchers have also observed altered platelet function in depressed patients, further increasing the risk for a thrombosis formation [75–77]. Patients with depression have been shown to have increased levels of the pro-inflammatory cytokines TNF-α, interleukin-1, and interleukin-6 [75–77]. The overproduction of these cytokines leads to inflammation eventually causing left ventricular remodeling and contractile dysfunction [77].
Sleep plays an important part in the maintenance of normal cardiac function. Sleep abnormalities, such as sleep apnea, constitute another CNS disorder that, similarly to depression, can play a role in the development and progression of HF. Sleep disordered breathing (SDB), often seen in obstructive sleep apnea (OSA), is associated with an increase in morbidity and mortality in HF patients [78, 79]. The most common manifestations of OSA are respiratory abnormalities, but research has revealed a strong relationship between OSA and cardiovascular disease such as arrhythmias [80], hypertension, coronary artery disease, and HF [81, 82]. The pathophysiological relationship between OSA and HF includes oxidative stress, sympathetic activation [83], systemic inflammation, and endothelial dysfunction [84]. The physiological relationship between OSA and decline in cardiac function is further illustrated through the improvement of cardiac function shown in patients with OSA treated with nocturnal continuous positive airway pressure (CPAP) treatment. Patients treated with CPAP were shown to have an increase in left ventricular ejection fraction and significant decreases in daytime systolic blood pressure and heart rate [85].
During normal sleep, distinct physiologic changes in cardiovascular function take place. This includes a decrease in sympathetic nerve stimulation, heart rate, blood pressure, cardiac output, and systemic vascular resistance (SVR) [86]. OSA occurs when the upper way becomes partially or completely closed during sleep causing either partial or complete lack of airflow [87]. This increased airway resistance leads to altered sleep and awakening patterns which ultimately result in significant changes in cardiac function. Studies have identified the mechanisms of OSA resulting in cardiovascular disease as four factors, including a prolonged exaggeration of negative intrathoracic pressure, development of hypoxia and hypercapnia during apnea, repetitive arousal from sleep at the end of apneic episodes, and altered endothelial function or vascular wall injury [88]. Extreme decreases in intrathoracic pressure are seen in patients with OSA. This decrease in pressure causes an increase in left ventricular transmural pressure, which leads to an increase in cardiac afterload and impaired left ventricular relaxation [86–89]. The blockage of the upper airway in OSA causes a hypoxic and hypercapnic state. The recognition of this state of decreased oxygenation causes intermittent states of arousal to take a breath [81]. These episodes of brief wakefulness cause an increase in myocardial oxygen demand [89]. When awake, there are bursts of sympathetic activity, which result in increased blood pressure and heart rate [86–89]. Blood pressures as high as 200/120 have been recorded post apneic episodes in patients suffering from OSA, and it is thought that this process occurring several times per night leads to hypertrophy of smooth muscle and increased peripheral resistance [81]. The repetitive reoxygenation from a hypoxic state also leads to the production of free radicals. The increase of reactive oxygen species (ROS) contributes to endothelial damage through oxidative stress [81, 86, 90]. This stress exacerbates the inflammatory response leading to an increase in inflammatory cytokines and endothelin. Increased levels of pro-inflammatory cytokines have been observed in patients with OSA [90]. The ensuing inflammation is associated with endothelial damage, increased arteriole resistance, and atherosclerotic plaque formation which can worsen the progression of HF (see above and Refs. [82, 87]).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


