CONGESTIVE HEART FAILURE AND CARDIOMYOPATHY
CHAPTER
Pathophysiology of Congestive Heart Failure
DEFINITION OF HEART FAILURE
There are multiple definitions of heart failure, but it is fundamentally a clinical syndrome. Similar to anemia or acute renal failure, heart failure is not a “standalone” diagnosis, but rather always possesses an etiology In some cases, however, the etiology cannot be determined. Virtually any form of heart disease can lead to heart failure. In general, heart failure is defined as a clinical syndrome characterized by shortness of breath and fatigue at rest or with exertion in the presence of underlying structural and/or functional heart disease. In advanced cases, salt and water retention are manifested by edema and organ dysfunction.
DEMOGRAPHICS OF HEART FAILURE
Heart failure is a common condition in the United States affecting more than 5 million people, with an overall self- reported prevalence of 2.4%. The incidence increases with age. At the age of 40, the lifetime risk of developing heart failure is one in five. In those older than 65, 10 per 1,000 people will have heart failure. The most common preceding diagnosis is hypertension (75%). Not only is it common in the general population, but it is also associated with high morbidity and mortality. There has been a 164% increasing hospitalization rate over the past 15 years, now accounting for almost 1 million hospitalizations and approximately 3.5 million outpatient visits, annually Heart failure accounts for about 7% of all deaths due to cardiovascular disease. Those who are diagnosed also have a shorter survival; 50% of those diagnosed with heart failure die in 5 years. In-hospital mortality is as high as 5% to 8% (Roger et al., 2011).
The morbidity associated with heart failure in an individual affects both ambulatory and inpatient care. The average patient takes six medications. Seventy-eight percent of patients have at least two hospitalizations per year. Up to 30% are readmitted within 90 days of hospitalization (Schrier and Gheorghiade, 2011). In the Medicare population, 18% have heart failure, which translates to 6 billion dollars of Medicare payments. They average one hospitalization per year, with an average stay of 7 days. These patients use outpatient services often, with about 10 physical office visits per year (Schneider et al., 2009). The annual cost attributed to heart failure is approximately $46 billion (Schrier and Gheorghiade, 2011).
THE INDEX EVENT
The event may be obvious, such as a sudden loss of a large mass of contractile tissue (i.e., an acute myocardial infarction), or it may be completely silent, such as the early expression of a mutant gene. In many cases, such as familial cardiomyopathy and the onset of valvular heart disease or hypertension, heart failure occurs after a lengthy latency period, or it may develop acutely, such as from acute aortic insufficiency due to bacterial endocarditis. The index event could take the form of acute lymphocytic myocarditis and manifest as heart failure only many months or years later. There are infinite genetic and environmental influences, which is why the natural history of heart failure and the pace at which it unfolds is so variable among individual patients. Uncertainty about the index event or etiology also makes the prognosis for any individual patient unclear. Given the heterogeneity in the presentation of heart failure, the description includes the stage of heart failure from being at risk (stage A), asymptomatic with structural heart disease (stage B), symptomatic heart failure (stage C), and those with refractory heart failure (stage D) (Fig. 15.1; Hunt et al., 2009).
Identifying the underlying etiology of the heart failure is an important part of the evaluation as it may directly impact long-term prognosis. Common etiologies include:
 Hypertension
Hypertension
 Myocardial ischemia/infarction
Myocardial ischemia/infarction
 Genetic (familial or hereditary)
Genetic (familial or hereditary)
 Diabetes mellitus
Diabetes mellitus
 Myocarditis (infectious, giant cell)
Myocarditis (infectious, giant cell)
 Infiltrative or restrictive (hemochromatosis, amyloidosis, sarcoidosis, Fabry disease)
Infiltrative or restrictive (hemochromatosis, amyloidosis, sarcoidosis, Fabry disease)
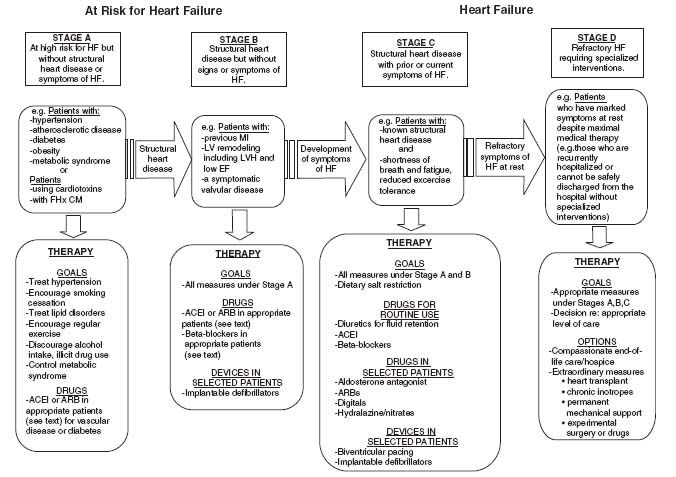
FIGURE 15.1 Stages in the development of heart failure/recommended therapy by stage. (Redrawn from Hunt SA, Abraham WT, Chin MH, et al. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration with the International Society for Heart and Lung Transplantation. J Am Coll Cardiol. 2009;53(15):e1-e90, with permission from Elsevier.)
 Substance use (alcohol, ephedra, cocaine)
Substance use (alcohol, ephedra, cocaine)
 Peripartum cardiomyopathy
Peripartum cardiomyopathy
 Connective tissue disease
Connective tissue disease
 Doxorubicin and other chemotherapy-induced cardiotoxicity
Doxorubicin and other chemotherapy-induced cardiotoxicity
 Infectious—Chagas, HIV Echovirus, Coxsackie
Infectious—Chagas, HIV Echovirus, Coxsackie
 Valvular heart disease
Valvular heart disease
 Other miscellaneous pathologies (e.g., pericardial disease)
Other miscellaneous pathologies (e.g., pericardial disease)
ADAPTIVE RESPONSES TO THE HEART FAILURE SYNDROME
The circulation adapts to a perceived disruption in homeostasis with both short-term and long-term adaptations. Shortterm adaptations include activation of the Frank-Starling mechanism and activation of the sympathetic nervous system (SNS). Long-term adaptations include heightened and alterations in the size and the shape of the heart (the so-called left ventricular [LV] remodeling). Although these adaptations may be somewhat protective in the short-term, over time they become counterproductive and contribute importantly to the pathogenesis of heart failure.
The Frank-Starling mechanism acts to increase the force of heart muscle contraction in response to an increase in end-diastolic volume (Fig. 15.2). In heart failure, however, this response is blunted, both at rest and during exercise. The force-frequency response is also attenuated in the failing heart, secondary to decreased norepinephrine (NE) stores and β-receptor density, which produces a decreased inotropic response to exercise so that less contractile force is generated in response to an increase in heart rate. Patients with heart failure can still call on the Frank-Starling mechanism, albeit at a reduced operational level. The inability to raise the stroke volume during exercise may be one of many reasons why patients have reduced exercise
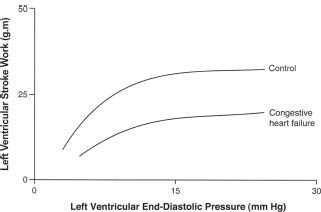
FIGURE 15.2 Heart failure is characterized by a diminished ability to increase the cardiac output or cardiac work in response to an increase in preload-Starlings law of the heart.
AUTONOMIC NERVOUS SYSTEM DYSFUNCTION
The SNS is activated early in the syndrome of heart failure, before overt signs and symptoms occur. Elevated plasma NE levels are observed and are an important marker of a poor prognosis. The mechanism that activates the SNS in heart failure is unknown. Increased local levels of synaptic NE in the heart increase the force of contraction and heart rate, offering early support for the failing heart. But this may also be the source of dysrhythmias and likely is responsible for the downregulation of β-adrenergic receptors. The failing heart tissue is itself also relatively depleted of NE, thus rendering the heart less responsive to sympathetic stimulation. There is less myocardial reserve in response to inotropic stimulation. The SNS also drives some of the increase in myocyte size, thus contributing to the LV remodeling process. Finally, the SNS activates the RAAS via β-receptors in the kidney, adding further to heightened peripheral resistance, salt, and water retention, and LV remodeling. In summary, early activation of the SNS in heart failure is “protective” by increasing heart rate, force of contraction, myocardial mass, and by protecting blood pressure, but there is a price to pay in the long run. Ultimately, excessive SNS activity is directly toxic to the heart and contributes importantly to the pathogenesis of heart failure.
Reflex control mechanisms are abnormal in heart failure (Fig. 15.3). Peripheral vascular resistance is increased, and there is defective parasympathetic control, an abnormal response to orthostasis, a blunted heart rate response to exercise and to pharmacologic vasodilation, impaired heart rate recovery from exercise, reduced heart-rate variability, and altered baroreceptor function. These abnormalities may improve following heart transplantation, suggesting that these are functional and not structural changes. They are rarely normalized. The precise cause of these abnormal reflex control mechanisms is not clear, but they may be the result of evolutionary forces that are acting to redistribute blood flow to more vital organs.
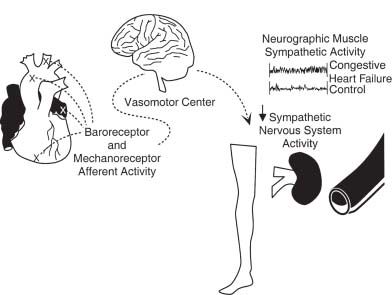
FIGURE 15.3 Baroreceptor and mechanoreceptor activation occurs when the heart is distended due to volume overload. This signal is processed by the brain and, in the setting of heart failure, fails to reduce sympathetic activity (the normal response). The result is enhanced sympathetic traffic to the periphery, vasoconstriction, and reduced renal blood
RENIN–ANGIOTENSIN–ALDOSTERONE SYSTEM
The RAAS is active in the circulation and in the tissue in heart failure. Probably 90% of the activity of the RAAS is embedded in the various tissues, including the heart, brain, and vasculature. This system, in conjunction with the SNS, plays a key role in the pathogenesis of the syndrome.
The RAAS is known to be activated by numerous mechanisms:
 Volume contraction
Volume contraction
 Low cardiac output
Low cardiac output
 Decreased renal blood flow
Decreased renal blood flow
 Hyponatremicperfusate to the macula densa
Hyponatremicperfusate to the macula densa
 β-Adrenergic stimulation to the kidney
β-Adrenergic stimulation to the kidney

 Salt and water restriction
Salt and water restriction
Angiotensin-II (Ang II) is a small, potent peptide produced by the cleavage of Ang-I by angiotensin-converting enzyme (ACE). Ang-II has a vast array of biologic activities, most of which contribute importantly to the pathogenesis of heart failure:
 Vasoconstriction
Vasoconstriction
 Vascular and cardiac myocyte growth, hypertrophy
Vascular and cardiac myocyte growth, hypertrophy
 Activation of fibroblasts with increased collagen production
Activation of fibroblasts with increased collagen production
 Facilitation of NE release
Facilitation of NE release
 Stimulation of aldosterone release
Stimulation of aldosterone release
 Volume expansion
Volume expansion
 Thirst stimulation
Thirst stimulation
 Arginine vasopressin release
Arginine vasopressin release
 Proinflammatory activity
Proinflammatory activity
 Direct toxicity to the myocardium when present in excessive quantities
Direct toxicity to the myocardium when present in excessive quantities
 Mesangial hypertrophy in the kidney
Mesangial hypertrophy in the kidney
 Increased intraglomerular hydraulic pressure via postglomerular efferent arteriole vasoconstriction
Increased intraglomerular hydraulic pressure via postglomerular efferent arteriole vasoconstriction



