Fig. 2.1
Mechanism of heart failure progression. The steps in the progression of heart failure
Various homeostatic mechanisms are activated in CHF, such as the activation of the renin-angiotensin-aldosterone system. Decreased renal perfusion is detected by receptors in the renal arterioles, leading to renin release from the kidneys. The increased angiotensin II that is formed acts on efferent arterioles increasing glomerular filtration pressure, despite reduction in renal perfusion pressure. Aldosterone synthesis is stimulated by angiotensin II, causing retention of salt and water by the kidney. Initially, this mechanism works to preserve normal systemic and renal perfusion. However, over a longer period this process leads to edema, elevated pulmonary artery pressures, and increased after-load [17]. Figure 2.2 illustrates these mechanisms.
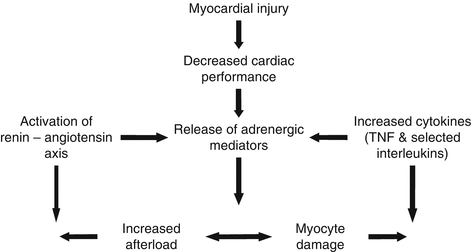
Fig. 2.2
Cycle of HF Progression, interacting with the renin-angiotensin axis, Adrenergic system and cytokines. Injury causing activation of the renin-angiotensin-aldosterone system, release of adrenergic mediators
Figure 2.2 shows that there is an increase in sympathetic activity, as well as vasopressin and renin release, in CHF. Neuro-endocrine activation is manifested by the release of norepinephrine, vasopressin and atrial natriuretic peptide [18]. The increased after-load and myocyte damage leads to a repetitive cycle of decreasing cardiac performance. Norepinephrine increases systemic vasoconstriction, chronotropy and inotropy by direct cardiac stimulation [19]. Over time, this increase in tissue norepinephrine activity predisposes to ventricular arrhythmias and sudden cardiac death. Higher circulating levels of plasma norepinephrine have been shown to negatively correlate with the prognosis of heart failure [20]. These values, while a rough guide, can be highly variable and unreliable compared to local concentrations of norepinephrine within the heart. The levels of norepinephrine in the cardiac circulation are measured by norepinephrine spill-over, and are elevated in CHF, providing an accurate prognostic guide [21].
Increased levels of endothelin are also found in CHF, promoting peripheral vasoconstriction, myocyte hypertrophy, and adverse remodelling [22]. Furthermore, atrial and brain natriuretic peptides (ANPs & BNPs) are released by the atria in response to stretch and increased atrial pressure. Neurohormones, cytokines and nitric oxide all interact together in complex ways to create the syndrome of chronic heart failure.
Fetal Gene Activation during Heart Failure
In a hypertrophied heart myocytes are not only big in size, but they also have additional sarcomeres. The manner in which these sarcomeres are added depends upon the type of load being imposed on myocytes. In situation of pressure overload, sarcomeres are added in parallel, leading to increased LV wall thickness, whereas, during volume overload, they are added in series, resulting in dilation of the ventricular cavity. There are also qualitative differences in sarcomeres of hypertrophied myocytes. A great body of evidence suggest that hypertrophy of myocytes is associated with induction of a group of genes, which are usually expressed during fetal heart development. These genes include activation of β-myosin heavy chain (MHC), skeletal-α-actin, atrial natriuretic factor and the repression of α-MHC and SR-CaATPase (SERCA). These changes may be initially salutary for an overloaded heart; however, prolonged hypertrophy leads to myocyte dysfunction, which eventually results into heart failure. Results collected from animal studies have also indicated that, the loss of α-MHC content is a critical determinant of the reduced myocardial contractility during heart failure. Direct evidence in support of a causal link between the loss of α-MHC and the development of heart failure came from experiments in which the α-MHC gene was ablated. Data correlating even small decrements of α-MHC content with changes in the intrinsic contractile characteristics of the myocardium also support the idea that a critical level of α-MHC content is essential for the normal pump function of the heart.
Another possible cause of a decrease in contractile protein-function is an alteration in the expression and/or activity of regulatory proteins. In animal models of heart failure, there are changes in the myosin light chain and the troponin-tropomyosin complex. Changes in myosin light-chain isoforms have been observed in heart samples of patients subjected to increased mechanical stress, and the expression of troponin-T splice variants was found to be altered in failing human myocardium. Changes in the phosphorylation status of Troponin-I have also been suggested to be involved in the loss of contractile activity of myocytes during heart failure. Moreover, defects in sarcoplasmic reticulum Ca2+ATPase and Ca release channels have been suggested to be responsible for the contractile dysfunction of myocytes.
Myocyte Cell-Death during Heart Failure
It has been realized that in addition to muscle gene dys-regulation, cardiomyocyte cell-death significantly contributes to loss of ventricular function in a failing heart. During increased workload, myocytes undergo a hypertrophic growth response to compensate for the increased demand. The initiating events of which are similar to those that drive cell-cycle progression in proliferating cells. A continuous growth signal in myocytes, at some point, causes cells to malfunction and leads to cell-death. As cells die, the workload on the remaining cells increases, which further aggravates this process and eventually leads to organ failure. Both in humans and animals with different cardiac disorders, including ischemic heart disease, idiopathic dilated cardiomyopathy, hypertensive heart disease, viral myocarditis, pacing-induced cardiomyopathy and different transgenic models of heart failure, myocyte cell-death has been implicated as a common cause of the cardiac pathology. However, the mechanism of cell death in a failing heart remains highly disputed. Some studies have documented a role of caspases in myocyte cell-death, however, other have disputed this notion. The activation of caspases in ischemic heart disease seems fairly accepted, but its participation in non-ischemic diseases remains controversial. Our own studies have shown that in pressure overloaded hearts PARP is not cleaved, a marker of caspase-dependent cell-death, but rather its expression is progressively increased in relation to the degree of cardiac hypertrophy, suggesting that hemodynamic-stress endangers cardiac myocytes through a mechanism that appears different from the conventional caspase-mediated apoptosis. Activation of PARP has been seen during different animal models of heart failure as well as in human failing hearts. Recent evidence suggests that prevention of cardiomyocyte cell-death by PARP-inhibition and/or by changing activity of other cell-death intermediate protects the overloaded heart from going into failure. G-protein coupled beta receptors, which have been implicated in the normal and abnormal functioning of cardiac muscle cells in response to beta-receptor signalling. These receptors and their mechanistic properties have recently been acclaimed by the Nobel committee that saw it fit to award the Nobel Prize in Chemistry for 2012 to Drs Lefkowitz and Kobilka [23].
Genetics of Heart Failure
Before we proceed to a discussion of the complex clinical manifestations of CHF, a brief discussion of the genetic factors at work in CHF is necessary for completion. Almost 20 % of patients with dilated cardiomyopathy are likely to have an inherited genetic defect. For instance, familial hypertrophic cardiomyopathy is caused by a number of gene mutations affecting beta myosin heavy chain, cardiac troponin T & I, alpha tropomyosin, myosin-binding protein C, and myosin light chains 1 and 2 [24]. In certain conditions such as Duchenne muscular dystrophy, there are definite genetic associations of an abnormal gene called dystrophin with abnormal myocardial function [25].
Cytoskeletal Proteins
Discoveries made during the last 20 years have revealed a genetic origin in many cases of dilated cardiomyopathy (DCM). Currently, over 40 genes have been associated with the disease. Mutations in DCM-causing genes induce the condition through a variety of different pathological pathways with complex and not completely understood mechanisms. Genes that encode for sarcomeric, cytoskeletal, nuclear membrane, dystrophin-associated glycoprotein complex and desmosomal proteins are the principal genes involved. In this review we discuss the most frequent DCM-causing genes. A classification has been proposed, in which DCM genes are considered as being major or minor genes according to their mutation frequency and the available supporting evidence [26].
Hypertrophic cardiomyopathy (HCM) is the most common monogenic genetic cardiac disease, with an estimated prevalence of 1:500 in the general population. Clinically, HCM is characterized by hypertrophy of the left ventricle (LV) walls, especially the septum, usually asymmetric, in the absence of any cardiac or systemic disease that leads to a secondary hypertrophy. The clinical course of the disease has a large inter- and intrafamilial heterogeneity, ranging from mild symptoms of heart failure late in life to the onset of sudden cardiac death at a young age and is caused by a mutation in one of the genes that encode a protein from the sarcomere, Z-disc or intracellular calcium modulators. Although many genes and mutations are already known to cause HCM, the molecular pathways that lead to the phenotype are still unclear [27].
Abnormal Lubrication in Diastolic Heart Failure
By regulating migration, proliferation and apoptosis as well as extracellular matrix synthesis and assembly, proteoglycans, integrins and the dystrophin-glycoprotein complex may be of great importance both during development and in vascular disease [28].
Syndecan-1 is a member of the proteoglycan family involved in cell-matrix interactions. In patients with heart failure, syndecan-1 levels correlate with fibrosis biomarkers pointing towards a role in cardiac remodeling. Studies have shown that this proteoglycan is elevated in patients with HF with preserved ejection fraction (diastolic dysfunction) [29].
Clinical Spectrum
Heart failure is usually defined as the inability of the heart to generate an output sufficient to meet the metabolic requirements of the body. The left or right ventricles may fail individually or together. Heart failure may also occur in the face of normal systolic ventricular function.
Left heart failure presents with shortness of breath which, if severe, will occur at rest. Patients are graded on their functional capacity depending on the level of activity that induces shortness of breath. This functional classification, known as the ‘New York Heart Association (NYHA) classification,’ is widely accepted and used around the world. This functional classification is used in the assessment of patients, prognostication and in tailoring management. The classes are as follows:
NYHA class I | Shortness of breath on unaccustomed exertion. Normal exercise tolerance |
NYHA class II | Shortness of breath on accustomed exertion, e.g., Breathlessness on walking uphill or climbing a flight of stairs |
NYHA class III | Shortness of breath on mild exertion, e.g., Symptomatic on walking a few metres |
NYHA class IV | Symptomatic at rest or on minimal exertion |
Minor impairment of cardiac function may remain asymptomatic but, as compensatory mechanisms become maladaptive, the clinical features of heart failure emerge. These relate primarily to the consequences of elevated atrial pressures and reduced cardiac output, expressed as congestion and peripheral hypoperfusion respectively. Manifestations of left and right heart may occur separately, but in reality they often occur together in varying degrees, resulting in the broader syndrome of Congestive Heart Failure (CHF).
Acute Left Heart Failure
This is usually caused by acute myocardial infarction, but may also be due to acute aortic or mitral regurgitation, or fulminant myocarditis. The patient usually presents with sudden onset of breathlessness and may cough up pink, frothy sputum. These clinical features are hallmarks of acute pulmonary edema although, in reality, most patients are now diagnosed on the basis of ‘wet lungs’ using chest x-ray. Systemic hypoperfusion supervenes, progressing to hypotension, oliguria and, in severe cases, cardiogenic shock.
Chronic Left Heart Failure
Patients usually have varying degrees of dyspnoea on exertion, the causes of which are many. For example, the elevation of left atrial pressure with exertion may be caused by:
Respiratory muscle fatigue
Metabolic factors such as acidosis and renal impairment, or
Muscle fatigue due to low cardiac output and poor muscle conditioning.
Clinical examination may reveal signs of low cardiac output and reflex sympathetic stimulation which include, tachycardia, cool peripheries and, occasionally, cyanosis. Auscultation may reveal inspiratory crackles at lung bases, although this can be unreliable; they are just as likely to be retained secretions within the lungs. Pleural effusions may be present. A third heart sound may be present producing a gallop rhythm. In severe heart failure, dilatation of the cardiac base and fibroskeleton causes mitral annular dilatation and mitral regurgitation. This is manifested by a pan-systolic murmur at the apex.
Acute Right Heart failure
Acute right heart failure may be caused by pulmonary embolism or right ventricular infarction. Patients usually present with breathlessness, systemic hypotension, cool skin, elevated jugular venous pressure and occasionally an enlarged liver.
Chronic Right Heart Failure
In this condition patients complain more of fatigue, and breathlessness is common. They also complain of a bloated feeling in the abdomen and a loss of appetite. This may be due to ascites, liver congestion, and edema of the gastro-intestinal tract. On examination the jugular venous pulse (JVP) is elevated. Occasionally there may be large ‘v’ waves in the JVP along with pulsatile hepatomegaly, suggestive of functional tricuspid regurgitation. In such patients a pan-systolic murmur can be heard at the left sternal edge. Peripheral edema and ascites may be present. Occasionally, patients manifest and present with jaundice and impaired protein synthesis, due to chronic impairment of liver function. Right heart failure is more commonly seen in the congenital heart disease population and patients with pulmonary hypertension. The increased use of long term left ventricular assist devices, has uncovered a group of sub-optimally supported failing right ventricles with interesting consequences.
Complications
A variety of cardiac arrhythmias occur in heart failure, especially atrial fibrillation, which causes varying degrees of haemodynamic deterioration. Ventricular arrhythmias occur later in the course of the disease and are more sinister, often causing sudden death. Arrhythmias and cardiac dilatation predispose to thrombus formation within cardiac chambers, which can then embolise into the pulmonary or systemic circulations. Chronic congestion of the lungs also provides a fertile ground for chest infections. Deep vein thromboses as a result of sluggish flow in the veins of the legs and pelvis may progress to pulmonary embolism and sudden death.
Advanced heart failure also causes progressive failure of major organs such as the liver and kidneys.
Diagnosis
As described earlier, CHF can present with a variable clinical picture. However, a variety of investigations can help confirm the diagnosis, such as Electrocardiogram (ECG), Chest X-ray, Echocardiography, Cardiac Catheterization, Radionuclide ventriculography (RNVG), Magnetic Resonance Imaging (MRI), and Cardio Pulmonary Exercise Testing. Great strides have also been made in the use of bio-markers in heart failure, such as BNP (brain related natriuretic peptide), and n-terminal Pro BNP (NT-BNP). Such investigative tools are useful for the mapping the course and progression of the disease. More recently, greater use of monitoring response to therapy with NT-BNP is gaining traction.
Current Treatment Options for CHF
Congestive heart failure is a complex medical condition requiring therapeutic interventions at multiple levels. It is the most common cause of medical admissions in Australia, the United States, Canada and the UK. The biggest advance in the treatment of heart failure has been the establishment of ‘Heart Failure Clinics’ and heart failure groups in hospitals, serviced by people interested in investigating, understanding and managing heart failure [30].
Multi-disciplinary heart failure programs that run cardiac rehabilitation courses after myocardial infarction or cardiac surgery, have been effective in reducing heart failure related admissions [31]. Symptomatic improvement has also been noticed, along with reduced hospitalisation in patients with cardiomyopathy who attend a specialised clinic [32].
Prevention also plays a very important role. The West of Scotland Study showed that primary prevention, by advocating drug therapy with pravastatin, prevented myocardial infarction and reduced the incidence of heart failure in a population at risk [33]. The 4S Study (Scandinavian Simvastatin Survival Study) followed patients with coronary artery disease, some of whom were randomly assigned to a placebo group and some to a group treated with simvastatin. Secondary prevention in the 4S Study showed that occurrence of heart failure at 5-year follow-up was significantly higher in the placebo than in the simvastatin group [34].
Exercise programs to improve general fitness and the level of exercise tolerance have been shown to significantly improve the functional status of patients with heart failure [35]. Bed rest, which was a cornerstone of traditional therapy for CHF, is therefore no longer advocated for these patients.
Despite these improvements in treatment and prevention the effective management of heart failure continues to be a pressing concern. Before discussing my research into the utility and efficacy of ventricular containment, this next section will review current approaches to the treatment of CHF, both medical and surgical.
Drug Therapy
Inotropic Agents
Digoxin, a drug that exerts a direct inotropic effect on the heart, has been a therapeutic mainstay in CHF for many years. Despite this the DIG trial (Digitalis Investigation Group) found that there was no significant impact on mortality in patients treated with digoxin [36]. However, this study demonstrated that patients treated with digitalis required less hospitalisation for worsening heart failure. The newer inotropic agents have also failed to lower the mortality rate of CHF sufferers. The PROMISE (Prospective Randomised Milrinone Survival Evaluation) trial, for example, did not live up to its name. Indeed there was a 28 % higher all cause mortality in patients randomly treated with 40 mg/day of milrinone compared to the placebo group [37].
Vasodilators
The first Veteran Administration Co-operative Vasodilator-Heart Failure Trial (V-HeFT 1) was published in 1986. The report showed that the addition of the vasodilators hydralazine (300 mg/day) and isosorbide dinitrate (160 mg/day) to a digoxin and diuretic regime resulted in a reduction in mortality of 38 % after 1 year, 25 % after 2 years, and 28 % over the entire follow-up period (a mean of 2.3 years) [38]. In 1991 the V-HeFT II study (conducted by the same group) reported that the ACE inhibitor enalapril was 18 % better than the combination of the two direct vasodilators [39]. However, the hydralazine-isosorbide combination was associated with improved ventricular function and better oxygen consumption at peak exercise.
Calcium Channel Blockers
Calcium channel blockers have been associated with worsening heart failure and a rise in mortality [40] with the exclusion of amlodipine. The PRAISE trial (Prospective Randomised Amlodipine Survival Evaluation) found that while amlodipine had no impact on mortality, it diminished the combined risk of non-fatal and fatal events by 31 % in patients with non-ischaemic cardiomyopathy [41].
Beta-Adrenoceptor Blockade
The sympathetic system is activated in patients with CHF. Another way of dealing with the adverse effects of neurohumoral activation is through beta-adrenoceptor blockage. Carvedilol, for example, is a non-selective beta-receptor antagonist that also blocks alpha-1 receptors and has antioxidant effects. In a recent trial carvedilol reduced mortality, 6–12 months after treatment, by 65 % in patients with CHF [42]. An Australian trial also showed improvement in ejection fraction, cardiac mortality and morbidity in patients treated with carvedilol [43].
Angiotensin-Converting Enzyme (ACE) Inhibitors
There is now a large volume of work supporting the role of ACE inhibitors in heart failure and these agents have become a cornerstone of first line therapy. Treatment with ACE inhibitors improves haemodynamic profiles as well as functional status, with benefits in exercise performance, dyspnea, fatigue and edema. The Survival and Ventricular Enlargement (SAVE) trial showed improvement in all cause mortality and reduced cardiovascular morbidity in patients with asymptomatic LV dysfunction after MI, who were treated with captopril [44]. The SOLVD (Studies of Left Ventricular Dysfunction) trial showed that enalapril reduced the risk of death and hospitalisation for worsening failure compared to the placebo group [45].
There are many other studies showing the efficacy of various ACE inhibitor drugs in heart failure as a consequence of MI. However, while most of these drugs delay progression of heart failure for a while and provide a solid bedrock of therapy, each of them has a significant incidence of complications. Despite best medical therapy, patients tend to worsen gradually as the ventricle dilates inexorably.
Surgical Therapy
Apart from first-line treatment with drug therapy, a number of surgical techniques have also been developed to treat heart failure. These include:
Ventricular Assist Devices
Mechanical circulatory support has finally come off age. We now have access to easy to deploy pumps such as the CentriMag (Thoratec Corporation) and CardioHelp (Maquet Cardiovascular) that can be used to salvage patients with acute heart failure and decompensation. In the more chronic heart failure patients, the use of left ventricular assist devices such as Heartmate II, Heartware and DuraHeart have made a dramatic difference to outcome. In a small group of patients with cardiomyopathy due to either idiopathic or to reversible causes, ventricular assist devices may be used on a long-term basis until the patients recover [46]. Despite many favourable reports [47] use of these devices were historically bedevilled by a high rate of bleeding, infection and hemolysis [48] as well as thrombo-embolism [49]. The present generation of continuous flow pumps work well in the short and intermediate term to tide patients over until transplantation [50]. However ventricular devices are expensive, require anti-coagulation and careful post-operative monitoring. Due to financial constraints, they are usually reserved for young patients in NYHA Class IV heart failure that are likely to be transplanted. The emerging area of destination therapy addresses patients who have terminal heart failure and are not candidates for transplantation. This field is growing rapidly in the US and almost 40 % of VADs are now for patients who are listed for Destination Therapy. In a sense, this is expensive therapy but is life-saving and prolonging in patients with terminal heart failure.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


