1 Pulmonary arterial hypertension (PAH)
1.1 Idiopathic
1.2 Heritable
1.2.1 BMPR2
1.2.2 ALK1, ENG, Smad9, Cav1, KCNK3
1.2.3 Unknown
1.3 Drug and toxin induced
1.4 Associated with
1.4.1 Connective tissue diseases
1.4.2 HIV infection
1.4.3 Portal hypertension
1.4.4 Congenital heart disease
1.4.5 Schistosomiasis
1.5 Pulmonary hypertension of the newborn
1′ Pulmonary veno-occlusive disease and/or pulmonary capillary haemangiomatosis
1″ Persistent pulmonary hypertension of the newborn (PPHN)
2 Pulmonary hypertension due to left heart disease
2.1 Systolic dysfunction
2.2 Diastolic dysfunction
2.3 Valvular disease
2.4 Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathies
3 Pulmonary hypertension due to lung disease and/or hypoxia
3.1 Chronic obstructive pulmonary disease
3.2 Interstitial lung disease
3.3 Other pulmonary diseases with mixed restrictive and obstructive pattern
3.4 Sleep-disordered breathing
3.5 Alveolar hypoventilation disorders
3.6 Chronic exposure to high altitude
3.7 Developmental abnormalities
4 Chronic thromboembolic pulmonary hypertension (CTEPH)
5 PH with unclear multifactorial mechanisms
5.1 Haematological disorders: chronic haemolytic anaemia, myeloproliferative disorders, splenectomy
5.2 Systemic disorders: sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis
5.3 Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders
5.4 Others: tumoral obstruction, fibrosis mediastinitis, chronic renal failure, segmental PH
Patients present with nonspecific symptoms, like breathlessness, fatigue, weakness, angina and syncope [3]. The New York Heart Association (NYHA) Functional Class (Table 45.2), based on clinical symptoms, is a strong predictor of survival [4, 5]. At physical examination, a left parasternal heave can be felt and auscultation may demonstrate an accentuated pulmonary component of the second heart sound, a pansystolic murmur of tricuspid regurgitation, a diastolic murmur of pulmonary insufficiency or a RV third sound. Different imaging techniques, including electrocardiography, chest radiography, echocardiography and cardiac magnetic resonance imaging, may raise the suspicion of the existence of PH, and these tests are also useful to identify possible underlying causes and to monitor treatment responses. Right heart catheterisation (RHC) is always needed to confirm the diagnosis, to evaluate the severity of the disease and to determine the effectiveness of drug therapy. The acute vasoreactivity test aids in determining drug therapy [2].
Table 45.2
Pulmonary hypertension New York Heart Association (NYHA) Functional Classification (FC) [75]
I | Patients with PH but without resulting limitation of physical activity. Ordinary physical activity does not cause undue dyspnoea or fatigue, chest pain or near syncope |
II | PH patients with slight limitation of physical activity. They are comfortable at rest. Ordinary physical activity causes undue dyspnoea or fatigue, chest pain or near syncope |
III | PH patients with marked limitation of physical activity. They are comfortable at rest. Less than ordinary activity causes undue dyspnoea or fatigue, chest pain or near syncope |
IV | PH patients with inability to carry out any physical activity without symptoms. These patients manifest signs of right heart failure. Dyspnoea and/or fatigue may even be present at rest. Discomfort is increased by any physical activity |
The current PH clinical classification gathers groups of PH that share similar haemodynamic criteria and types of pulmonary vascular lesions to optimise therapeutic approaches, predict patient outcomes and facilitate research strategies (Table 45.1) [1]. Group 1 PH corresponds to pulmonary arterial hypertension (PAH). PAH is characterised by precapillary PH (mPAP ≥25 mmHg, with a normal pulmonary capillary wedge pressure ≤15 mmHg) due to major pulmonary arterial remodelling. The lowest reported prevalence and incidence of PAH are 15 cases/million adult population and 2.4 cases/million adult population/year, respectively. The prevalence of PAH in Europe is estimated between 15 and 50 subjects/million population [6]. In 70 % of the heritable PAH cases, a germ line mutation of the bone morphogenetic protein receptor 2 (BMPR2) is found [7, 8]. The same mutation is found in 11–40 % of sporadic PAH patients, indicating the genetic predisposing factor for PAH [9].
In the year 2000, exonic mutations in the gene encoding for bone morphogenetic protein receptor type 2 (BMPR2) were found in 54 % of PAH patients, or more specifically 58–74 % of patients with heritable PAH (hPAH) and in 3.5–40 % of patients with sporadic PAH [8, 10–14]. BMPR2 is a member of the receptor family of transforming growth factor-β (TGF-β). The penetrance of mutations of BMPR2 is below 20 %, indicating that the BMPR2 gene is not the only gene responsible for PAH and that the pathophysiology of this disease is multifactorial. Therefore, many laboratories have investigated possible mutations in other members involved in the signalling cascade of TGF-β, and two genes were found mutated: ACVRL1 (activin A receptor type II–like kinase 1) and ENG (endoglin). However, these mutations account for only a small proportion of cases of hPAH. Recently, mutations have also been described in Smad 1, Smad 4, Smad 8 and Smad 9 [15, 16]. All these mutations disrupt the BMP/Smad signalling pathway and promote endothelial and smooth muscle apoptosis and proliferation, resulting in loss of the endothelial barrier function and pulmonary vascular remodelling. These mutations could also increase the susceptibility to inflammatory stimuli [17]. Recently, Ma et al. [18] demonstrated the involvement of the potassium channel subfamily K member 3 (KCNK3) missense mutations in the pathophysiology of PAH. Indeed, mutations in this gene were identified in six unrelated patients with PAH (three patients displaying heritable form of PAH out of 93 patients [3.2 %] and three patients with sporadic PAH out of 230 patients [1.3 %]) [18]. To date, all identified KCNK3 mutations are missense mutations and are responsible for a loss of function of the two-pore-domain potassium channel TASK-1 and its signalling pathway in PAH. The reduction in potassium channel activity may enhance calcium channel-mediated vasoconstriction and vascular remodelling [19, 20].
Pulmonary vascular remodelling, occurring mostly in the small- to midsized pulmonary arterioles (≤500 μm), is a hallmark of most forms of PH. This process is ascribed to the increased proliferation, migration and survival of pulmonary vascular cells within the pulmonary artery wall, i.e. pulmonary vascular smooth muscle cells (SMCs), endothelial cells (ECs), myofibroblasts and pericytes. PAH is associated with excessive production of vasoconstrictive mediators such as endothelin (ET)-1 concurrent with a reduced bioavailability of vasodilator molecules nitric oxide (NO) and prostacyclin (PGI2). Pulmonary vascular remodelling is also under the control of various key growth factors such as platelet-derived growth factor (PDGF), serotonin (5-hydroxytryptamine; 5-HT) and fibroblast growth factor (FGF)-2. Abnormalities in the expression and function of calcium and potassium channels are also involved in pulmonary vasoconstriction and remodelling of the pulmonary vasculature. Recent findings highlight the critical role of the close and complex relationship between the pulmonary vascular endothelium and inflammation/autoimmunity in PAH. Indeed, circulating levels of certain cytokines and chemokines are abnormally elevated, and some have been reported to correlate with a worse clinical outcome in patients with PAH [21–24]. Altered regulatory T (Treg) cell function has been demonstrated in patients with PAH, a phenomenon that has been demonstrated to be partly leptin-dependent [25, 26]. Similarly, natural killer (NK) cells have recently been implicated [27]. An accumulation of immature dendritic cells (DCs) has been demonstrated, suggesting that they may contribute to PAH immunopathology [28]. Furthermore, ectopic lymphoid follicles that develop in contact with remodelled pulmonary arteries could be the site of a local autoimmune reaction, leading to the production of autoantibodies directed notably against pulmonary vascular cells. Circulating autoantibodies are commonly detected in idiopathic PAH (iPAH) patients without evidence of an associated autoimmune condition [29–32]. However, despite the many arguments supporting a role of inflammation in the pathogenesis of PAH, only some patients respond to anti-inflammatory and/or immunosuppressive therapy. It is therefore necessary to understand the complexity of the immune mechanisms of PAH to improve the transfer of knowledge to the clinic.
Although PAH is still a disease without a cure, there are approved drug therapies that at least temporarily stabilise or improve the symptoms in the majority of patients. In this chapter, we will first outline the pathophysiological mechanisms that underlie PAH and PAH-associated RV failure. Subsequently, we will discuss the major therapeutic targets which are currently available or under development (Chap. 59).
45.2 Pathophysiology of PAH
The extensive structural and functional remodelling of the vasculature in lungs of patients with PAH takes place sequentially and includes medial hypertrophy, muscularisation of small arterioles, intimal thickening and the formation of plexiform lesions. These processes involve changes in all three layers (intima, media and adventitia) of the vessel wall and are the consequence of cellular hypertrophy, hyperplasia, inflammation, apoptosis, migration and accumulation of extracellular matrix (ECM).
45.2.1 Pulmonary Endothelial Cell Dysfunction
Pulmonary endothelial dysfunction is a critical element in the development and progression of PH, irrespective of disease origin. In PAH, the dysfunctional endothelium shows several abnormalities: (a) a transition from a quiescent state (having no adhesiveness) to an activated state, expressing specific markers and proteins, such as E-selectin and key adhesion molecules [e.g. intercellular adhesion molecule (ICAM-1) and vascular adhesion molecule (VCAM-1)] (submitted data); (b) a reduced ability to produce vasodilatory mediators such as NO and PGI2; (c) an excessive production and release of vasoconstrictive mediators such as 5-HT, ET-1 and Ang II [33]; (d) an important qualitative and quantitative remodelling of components of the ECM; and (e) an increased production of various factors affecting the control of proliferation, differentiation and migration of pulmonary vascular cells such as FGF-2 (basic), interleukin (IL)-6 and leptin [26, 34–36]. Furthermore, the pulmonary ECs derived from iPAH patients exhibit an aberrant cell phenotype which is characterised by an excessive proliferation and resistance to apoptosis induction [35, 37]. Tu et al. [35] have demonstrated that an excessive FGF-2 autocrine loop is one of the mechanisms involved in this aberrant endothelial phenotype, explaining the constitutive activation of the mitogen-activated protein kinase (MAPK) signalling pathway and the overexpression of two key anti-apoptotic factors B-cell lymphoma 2 (BCL2) and B-cell lymphoma-extra large (BCL-xL).
In PAH pulmonary ECs, many other intrinsic abnormalities were also described including p130cas overexpression, a key amplifier of receptor tyrosine kinase (RTK) downstream signals, altered energy metabolism and a constitutive activation of hypoxia-inducible factors (HIF)-1α [38, 39]. In addition, the abnormal cellular crosstalk between ECs and the other pulmonary vascular cells in the pulmonary vascular wall in PAH represent a key feature of PAH pathogenesis. We have shown that dysfunctional pulmonary ECs from patients with iPAH, through an aberrant release of FGF-2 and IL-6, contribute to increased pericyte coverage of distal pulmonary arteries in PAH, an abnormality that is a potential source of smooth muscle-like cells [36]. Indeed, activated TGF-β in pulmonary arterial walls in PAH can promote human pulmonary pericyte differentiation into contractile smooth muscle-like cells. Multiple lines of evidence therefore suggest that neutralisation of FGF-2, IL-6 and TGF-β1 may be beneficial against the progression of PAH. A better understanding of the underlying mechanisms is critical to slow down and reverse this obliterative pulmonary vascular remodelling in PAH. Experimental work strongly supports the fact that the obstructive vascular remodelling may be limited by strategies which, at a time, promote vasodilation and inhibit cell proliferation/survival and inflammation. Because many of these tools have been developed and are available through cancer treatment, there is a growing interest for the transfer of these tools to PAH. However, several studies are needed not only to identify the best strategies/molecules for use in PAH but also to better understand the risk/benefit of these anti-proliferative treatments, especially vis-à-vis the maintenance of cardiac function.
45.2.2 Pulmonary Smooth Muscle Hyperplasia
Mechanisms underlying the excessive pulmonary vascular SMC proliferation in PAH are partially understood and result from two complementary mechanisms: inherent characteristics and dysregulation of molecular events that govern SMC growth, including signals originating from pulmonary ECs. Cultured pulmonary arterial SMCs from patients with iPAH grew faster than SMCs from controls at basal conditions or when stimulated by 5-HT, FGF-2, epidermal growth factor (EGF), PDGF or fetal calf serum (FCS). For example, 5-HT transporter (5-HTT) activity is associated with pulmonary artery smooth muscle cell proliferation, and the L-allelic variant of the 5-HTT gene promoter, which is associated with increased expression of 5-HTT, is present in homozygous form in 65 % of patients with iPAH compared with 27 % of controls [40].
These observations explain the fact that interest has been growing in the potential use of anti-proliferative approaches in PAH [41]. Excessive release of various growth factors that are encrypted in the ECM and/or modification of growth factor production, receptor expression and/or alterations in the intracellular mitogenic signals have also been reported to contribute to this excessive smooth muscle migration, proliferation and survival. Inhibition of various RTK signalling pathways by specific inhibitors, such as imatinib, gefitinib and dovitinib, have been shown to exert beneficial effects in animal models of PH [34, 39, 42, 43]. However, further efforts still need to be made in order to establish the long-term safety and efficacy of these anti-proliferative approaches in PAH and their potential additive benefit with other drugs. Recent investigations also suggest that a chronic shift in energy production from mitochondrial oxidative phosphorylation to glycolysis (the Warburg effect) of pulmonary vascular cells is present and may participate in the pathogenesis [44–46]. Mechanistic studies focusing on cell metabolism and its interface with the genetic basis of PAH and inflammation are needed for a better appreciation of its role in the promotion of SMC proliferation and survival and to the disease progression.
45.2.3 Perivascular Inflammatory Cell Accumulation
In the past two decades, understanding of inflammation associated to PAH has moved from a common histopathological curiosity to a key pathomechanism that could be detrimental both in terms of disease susceptibility and development of pulmonary vascular remodelling. Histopathologically, pulmonary vascular lesions occurring in patients with PAH as well as in animal models of PH are characterised by varying degrees of perivascular inflammatory infiltrates, comprising of T and B lymphocytes, macrophages, DCs and mast cells. Recently, correlations were found between the average perivascular inflammation score and the intima plus media and adventitia thickness or mPAP, supporting a role of perivascular inflammation in the processes of pulmonary vascular remodelling [47]. In addition, inflammation precedes pulmonary vascular remodelling in animal models of PH, strongly supporting the notion that increased perivascular immune cell infiltration around lung vessels plays a key role in PAH development and progression [25]. As previously discussed, circulating levels of certain cytokines and chemokines are abnormally elevated and can directly control cell proliferation, migration and differentiation of pulmonary vascular cells.
There seems to be a particular role for IL-6 in the pathogenesis of PAH. Delivery of recombinant IL-6 protein in rodents is sufficient to cause pulmonary vascular remodelling and PH or to exaggerate the pulmonary hypertensive response to chronic hypoxia [48, 49]. Furthermore, IL-6-overexpressing mice spontaneously develop PH and pulmonary vascular remodelling, whereas IL-6 knockout mice are more resistant to the development of PH induced by chronic hypoxia [50, 51]. Recent data from our group demonstrated that the overabundance of macrophage migration inhibitory factor (MIF) plays a pivotal role in the pathogenesis of PAH. MIF is a critical upstream inflammatory mediator with pleiotropic actions partly explained by its binding to the extracellular domain of the endothelial CD74. In endothelial cells, activation of the CD74 can lead to activation of Src-family kinase and MAPK/ERK, PI3K/Akt and nuclear factor-kappa B (NF-κB) pathways and to apoptotic resistance by increasing the anti-apoptotic factors BCL2 and BCL-xL and by inhibiting p53 (submitted data). In addition, MIF can bind to C-X-C chemokine receptor type 2 (CXCR2) and type 4 (CXCR4), lead to the proliferation of pulmonary artery smooth muscle cells and contribute to hypoxic PH [52–54]. While this body of knowledge provides a preliminary understanding, it also highlights subtleties and complexities that require further investigation to determine whether anti-inflammatory strategies will be useful in PAH treatment in the future.
45.2.4 Impaired Pulmonary Angiogenesis
Multiple lines of evidence suggest that angiogenesis is clearly disturbed in experimental and human PAH with loss and progressive obliteration of precapillary arteries leading to a pattern of vascular rarefaction (“dead-tree” picture). However, high levels of different angiogenic factors including FGF-2 and VEGF are present in patients with iPAH, strongly supporting the notion that this phenomenon is probably due to signalling defects in the endothelium in PAH. Cool et al. demonstrated exuberant expression of the VEGF receptor KDR, coupled with a reduced expression of p27/kip1 (a cell cycle inhibitory protein) in the pulmonary ECs of plexiform lesions [55]. Since increased pericyte coverage in iPAH has been recently reported, another explanation might be related to abnormal pericyte recruitment or to intrinsic abnormalities in pulmonary pericytes in PAH [36]. A greater understanding of the role of pulmonary pericytes in vascular homeostasis and remodelling is needed.
45.2.5 In Situ Thrombosis
PAH pathological specimens often display thrombotic lesions in the absence of clinical or pathological evidence of pulmonary embolism, suggesting an in situ clotting phenomenon [56, 57]. In addition, PH is associated with a hypercoagulable phenotype that includes vascular upregulation of tissue factor and an increase in circulating levels of von Willebrand factor or plasma fibrinopeptide A [58–60].
45.3 Development of Right Heart Failure
Despite its meagre ability to respond to a rapid increase in pressure, the RV is usually able to adapt to a gradually increasing afterload by augmenting its contractility and wall thickness. The one metric which best describes RV adaptation in PAH is ventriculo-arterial coupling, which takes into account both contractility and afterload. When the increased afterload is matched by an adaptive increase in RV contractility and mass, the RV is said to be coupled to the pulmonary arterial circulation [61]. However, in the majority of patients with PAH, the severity and chronicity of the afterload increase ultimately overwhelm the increases in RV mass and contractility. The final course of PAH is therefore characterised by RV dilatation and failure, and eventually death (see Fig. 45.1). It has been speculated that, as in LV failure, neurohormonal activation may be central in the transition from RV adaptation to RV failure [63]. Indeed, sympathetic nervous system activity is increased in PAH patients, which finding has prognostic significance [64]. Chapters 5 and 6 explain more about the involvement of sympathetic and parasympathetic nervous systems in heart failure. Likewise, an increased renin–angiotensin–aldosterone system (RAAS) activity (described in Chap. 35) reflects PAH disease severity [65].
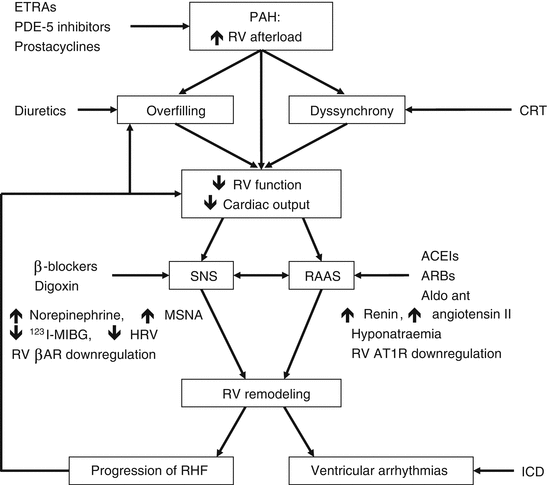
Fig. 45.1
Schematic overview of hypothetical pathophysiological mechanisms in PAH-related right heart failure, showing the multiple interactions between mechanical events (pressure overload, dilatation), electrophysiological changes (dyssynchrony, arrhythmias) and neurohormonal activation (Reproduced with permission from Ref. [62]). RV right ventricular, ETRAs endothelin receptor antagonists, PDE-5 phosphodiesterase-5, CRT cardiac resynchronisation therapy, SNS sympathetic nervous system, RAAS renin–angiotensin–aldosterone system, ACEIs angiotensin-converting enzyme inhibitors, ARBs angiotensin receptor blockers, MSNA muscle sympathetic nervous activity, HRV heart rate variability, βAR cardiomyocyte β1-adrenergic receptor, AT1R cardiomyocyte angiotensin type 1 receptor, Aldo ant aldosterone antagonist, RHF right heart failure, ICD implantable cardioverter defibrillator
A typical feature of RV failure is the prolongation of the systolic contraction time in comparison to the left ventricular contraction time, leading to a leftward shift of the septum at the end of RV contraction (during which time the LV is already in its relaxing phase) and impaired LV filling [66]. In addition to a systolic functional impairment, RV failure is characterised by diastolic dysfunction, which probably comes about through a combination of intrinsic stiffness and fibrotic replacement of RV cardiomyocytes [67, 68]. The transition from RV adaptation to RV failure is further characterised by reduced myocardial perfusion, which may not only reflect reduced coronary perfusion due to, e.g. systemic hypotension, but also an impairment in angiogenesis relative to the degree of hypertrophy [69, 70]. Metabolic remodelling is another recently highlighted characteristic of RV failure and includes a decreased uptake of fatty acids and an increased generation of ATPs through glycolysis rather than through glucose oxidation [71, 72].
45.4 Specific Drug Therapy
To facilitate a treatment plan, it is important to rule out all the possible underlying causes that could induce and cause progression of PAH. In addition to treating the underlying cause, when such a treatment is not available in the cases of idiopathic and heritable PAH, there are PAH-specific approved drugs which aim to dilate pulmonary vessels. In vitro and animal studies suggest that these drugs also have inhibitory effects on vascular remodelling [73]. Long-term treatment in patients has not led to a demonstrable regression of vascular remodelling [47]. Novel therapies in development (discussed later) show promising results with regard to inhibition of cell proliferation and inducing apoptosis, thereby limiting the progressive changes in morphometry [74].
45.4.1 Drugs Targeting the Pulmonary Vasculature
The currently available drugs for PAH treatment mainly target vasoconstriction via three biochemical pathways: ET-1, NO and PGI2 (Fig. 45.2) (see also Chap. 59) [76]. Experimentally, these therapies have some anti-proliferative effects [77, 78]. Table 45.3 shows FDA-approved therapeutics intervening in these three major pathways.
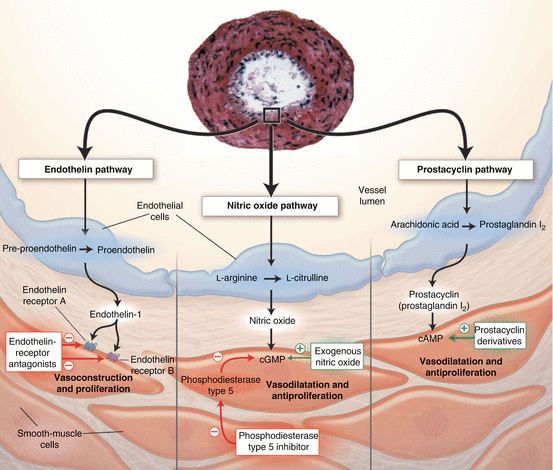
Fig. 45.2
The pathways involved in contraction and proliferation of pulmonary arterial smooth muscle cells and the endothelium. The four biggest groups of drugs available for PAH that target these pathways are endothelin receptor antagonists, nitric oxide, phosphodiesterase type 5 inhibitor and prostacyclin derivatives (Reproduced with permission from Ref. [75])
Table 45.3
FDA-approved drug therapies in PAH tested in randomised clinical trials
Drug | Study + reference | Improvement of | Serious adverse effects | ||||
|---|---|---|---|---|---|---|---|
6MWD/exercise capacity | Haemodynamics | FC | Survival | Time to clinical worsening | |||
CCB a | |||||||
Amlodipine, diltiazem, nifedipine | + | + | + | + | Systemic hypotension, bradycardia, oedema, headache, nausea | ||
NO/cGMP | |||||||
Riociguat | PATENT [80] | + | + | + | + | Hypotension, syncope | |
Sildenafil | + | + | + | Headache, flushing, epistaxis | |||
Tadalafil | + | + | + | Headache, flushing, epistaxis | |||
PGI2 | |||||||
Beraprost | +b | Headache, flushing, jaw pain, diarrhoea, approved in Japan/South Korea | |||||
Epoprostenol | + | + | + | Local site infection, catheter obstruction and sepsis (pump related) | |||
Iloprost (inhal) | + | + | + | + | Flushing, jaw pain | ||
Treprostinil | +d | + | Infusion site pain | ||||
ET-1 | |||||||
Ambrisentan | ARIES-1, ARIES-2 [100] | + | + | + | Peripheral oedema | ||
Bosentan | + | + | + | + | |||
Macitentan | SERAPHIN [106] | + | + | ||||
Combination | |||||||
Initial | Galie [107] | + | + | ||||
Sequential | + | + | |||||
45.4.1.1 Calcium Channel Blockers
Intracellular calcium levels are elevated in pulmonary arterial SMCs of PAH patients, leading to contraction of the muscular layer in the vessels. Less than 10 % of the patients with iPAH respond to an acute vasodilator, like inhaled NO or iloprost. This small group harbours patients who obtain improvement of symptoms after treatment with calcium channel blockers such as long-acting nifedipine, diltiazem or amlodipine (Chap. 37). The patients who respond to long-term calcium channel blocker therapy exhibit a more pronounced reduction in mPAP, reaching an absolute mPAP of 33 ± 8 mmHg with acute vasodilator testing. As a result, the consensus definition of a response is now defined as a fall in mPAP of ≥10 mmHg, to an mPAP ≤40 mmHg, with an unchanged or increased cardiac output. Patients with iPAH who meet these criteria may be treated with calcium channel blockers [79, 110]. Owing to potential negative inotropic effects, verapamil should be avoided [111]. If the patient does not respond well to the calcium channel blockers (CCB), medication directing the NO, PGI2 or ET-1 pathway should be added or replace the current treatment [77].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


