Figure 10–1.
Effect of an abnormally increased late Na+ current (late I Na ) to cause prolongation of the action potential duration and the QT interval in the electrocardiogram (ECG), and to cause early afterdepolarizations (EAD)
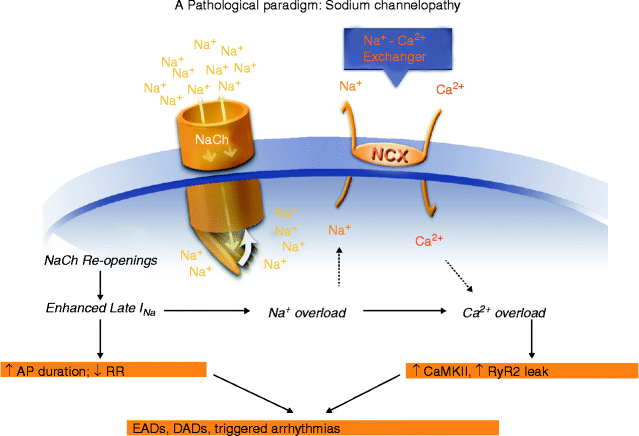
Figure 10–2.
Pathological consequences of acquired and/or inherited Na+ channelopathies
The Sodium Channel Late Current (Late INa)
Voltage-gated sodium channels (VGSCs) are heteromultimeric membrane pore-forming proteins whose conductance of Na+ is gated by changes in the membrane potential. VGSCs are required for the generation and propagation of action potentials (AP) in heart, nerve, and skeletal muscle. Sodium channel opening and peak INa are usually short in duration (often 1–2 ms) in these tissues, because Na+ channels quickly inactivate after opening. Sodium channels inactivate at depolarized potentials when the inner gate of the channel moves to block the pore. Upon membrane repolarization, Na+ channels rapidly revert to the closed state. Presumably the high numbers of charged residues in the voltage sensing domains of the channel render the closed state very energetically stable (this may be important for survival of the organism). However, the inactivated state(s) of the Na+ channel (which occur during prolonged depolarization) are not as stable, and Na+ channel reopening from an inactivated state(s) may occur. A small INa (late or sustained INa) therefore persists during prolonged depolarization such as the plateau (phase 2) of the cardiac AP [20–26]. Unlike skeletal muscle and neuronal cells, heart cells undergo prolonged depolarization during the plateau (phase 2) of every AP, and thus cardiac Na+ channels reside in inactivated states for approximately one-third of each cardiac cycle. Pathological conditions that lead to a failure of inactivation of Na+ channels are thus likely to cause a large increase of late INa and Na+ loading in the heart [27].
The functional relevance of cardiac late INa under physiological conditions is still unclear. Experimental [20, 28] and modeling studies [29] suggest that a relatively small persistent INa and few late openings of Na+ channels can significantly increase AP duration in the heart. However, the presence of larger late INa (relative to peak INa) in Purkinje fiber and M cells than in ventricular epicardial myocytes may contribute to the longer AP duration [22, 30–33] and greater propensity to occurrences of early after-depolarizations (EADs) in the former cells [31–33]. The transmural distribution of late INa appears to be species-dependent [26, 31–35].
Several mechanisms have been proposed to explain the presence of late INa, including window current [21, 36, 37] and specialized populations of Na+ channels (reviewed in [11, 15, 38]). An overlap of the voltage ranges wherein Na+ channels activate and inactivate (i.e., approximately −70 to −50 mV) can lead to a steady-state Na+ window current that will contribute to late INa. Steady-state TTX-sensitive Na+ window current at 35–37 °C in sheep [21] and rabbit [36] Purkinje fibers was found to occur from −70 to −20 mV, a range that is wider than predicted. The ratio of steady-state to peak INa was 0.06 % in rabbit Purkinje fibers [36]. One might expect window current to be most important during ischemia, and during phase 3 of the cardiac AP, when myocytes are depolarized to voltages at which window current is present. Because Na+ channel inactivation and activation as a function of voltage are altered by channel mutations [39], and perhaps by other channel modifications such as phosphorylation, the voltage range for Na+ channel “steady state” window current is not always clear. However, in the last 20 years, many researchers have recorded late INa and Na+ channel openings that decrease with time and at voltages that are putatively outside the commonly-accepted potential range of the Na+ window current (for example, voltages during the AP plateau are 0–20 mV, and outside the range for window current). The term “modal gating” (transient openings, scattered late openings and/or burst openings) has been applied to this form of Na+ channel activity [23, 25, 26, 29, 31, 40–45]. Evidence from studies of cardiac Nav1.5 currents [46, 47] suggests that late and peak INa are conducted by the same channels. However, neuronal Na+ channel isoforms including Nav1.1, Nav1.3 and Nav1.6 are also expressed in the heart [11, 48–53] and may contribute to late INa.
Causes of Increased Late INa: Inherited Na+ Channelopathies Causing Long QT (LQT) Syndrome 3
Mutations in the cardiac Na+ channel gene SCN5A cause LQT3. More than 80 LQT3 mutations have been identified to date [4]. Most are missense mutations and cause gain-of-function by increasing the probability that the cardiac channel NaV1.5 will fail to inactivate properly or will more readily re-open from a closed state to generate increased late Na+ current [37, 54–58]. A fivefold increase of late INa caused by the ΔKPQ mutation in NaV1.5 was found to greatly increase loading of the myocyte with Na+ in a modeling study by [27]. Many SCN5A mutations that enhance late INa are found in the DIII-DIV linker (including ΔKPQ), transmembrane segments DIVS4 and DIVS6, and the carboxy terminus of the channel, as befits their putative roles as “inactivation gate”, the voltage sensor whose movement generates the signal for movement of the inactivation gate, the inactivation gate receptor, and the “latch” which keeps the inactivation gate closed, respectively [4, 56]. Some of these mutations occur at sites that are also known targets for phosphorylation by protein kinases [59, 60].
Four different β-subunits, Navβ (β1 – β4), have been identified to play a critical role for cell surface expression, modulation of voltage-dependence, and transduction from cell signaling molecules to the pore-forming α-subunit of cardiac Na+ channels [61–67]. NaV1.5 channel α subunits in ventricular myocytes were reported to co-localize with β2 and/or β4 subunits at intercalated discs [50].
The congenital mutation L179F in the β4 subunit encoded by the gene SCN4B was reported to be responsible for the prolonged QT interval in a patient with 2:1 atrioventricular block [68]. Patch-clamp studies revealed a three- to eight-fold increase (gain-of-function) in late INa when L179F was co-expressed with an α-subunit. Recently, Ackerman and colleagues [69] reported a congenital mutation (V36M) in the β3 subunit encoded by the gene SCN3B that was associated with SIDS. Patch-clamp studies revealed a marked decrease (loss-of-function) of peak INa but a marked increase (gain-of-function) in late INa when V36M was co-expressed with an α-subunit. Similarly, mutations in the gene SCN1B encoding the β1 subunit have been shown to cause febrile seizures [70].
Causes of Increased Late INa: Inherited Mutations in Non-Channel Proteins as Causes of Long QT (LQT) Syndromes and/or Enhanced Late INa
Caveolae are a special type of lipid raft that in which receptors and signal transducing molecules are organized [71]. In the heart, caveolin-3 (encoded by the gene Cav3) is responsible for caveolae formation and is abundantly present in atrial, ventricular, and nodal cells [72]. Mutations in Cav3 can lead to LQT9. It has been shown that Nav1.5 is co-localized with caveolin-3 [73]. When the caveolin-3 mutations responsible for LQT9 in patients were heterologously expressed with Nav1.5, a two- to three-fold increase in late INa was observed. This increase in late INa was suggested to be the principal mechanism for proarrhythmia in these patients [74]. Consistent with this interpretation is the finding that caveolin-3 mutations were identified in samples from a cohort of sudden infant death syndrome (SIDS) patients [75].
The SNTA1-encoded α1syntrophin (SNTA1) is a member of the dystrophin-associated multiprotein complex that interacts with the PDZ domain-binding motif of the cardiac Na+ channel carboxyl terminus [76]. SNTA1 has been shown to interact with neuronal nitric oxide synthase (nNOS) and Ca2+/calmodulin-dependent ATPase (PCMA4b) [77]. Nitric oxide (NO) has been shown to increase the amplitude of late INa in rat ventricular myocytes [78] and in the presence of STNA1, PMCA4b acts as a potent inhibitor of NO production by nNOS. Patients identified with SNTA1 mutations [79–81] have been shown to have a prolonged QT interval (LQT12 syndrome) that may be associated with SIDS. Both peak and late INa were found to be increased when SNTA1 congenital mutant proteins were co-expressed with NaV1.5, nNOS and PMCA4b in cells.
Ankyrin-G is required for localization of NaV1.5 to the intercalated discs of cardiac myocytes [82, 83]. The protein βIV-spectrin binds to ankyrin-G and to CaMKII and thus targets CaMKII to the Na+ channel, where it phosphorylates S571 [83]. Because the phosphorylation of NaV1.5 by CaMKII leads to an increase of late INa [84–86], changes in the function of βIV-spectrin may lead to changes in late INa. Recently, defects in other intracellular proteins that are known to interact with VGSCs have been shown to enhance late INa. An excellent review of protein interactions with Na+ channels has been published recently [87].
Causes of Increased Late INa: The Acquired Na+ Channelopathies in Myocardial Ischemia and Heart Failure
An imbalance between myocardial oxygen supply and demand, as occurs during ischemia, heart failure, or tachycardia, leads to increases of myocardial concentrations of Na+ and Ca2+ [88, 89]. During ischemia and reperfusion following ischemia, amphiphilic lipids (e.g., palmitoyl-L-carnitine, lysophosphatidylcholine), glycolytic metabolites, reactive oxygen species (ROS) and catecholamines are increased, and intracellular concentrations of H+, Na+, and Ca2+ are elevated. Sodium-hydrogen exchange (NHE), reverse mode Na+-Ca2+ exchange (NCX with Ca2+ entry), and CaMKII activities are increased. Hypoxia, palmitoyl-L-carnitine, lysophosphatidylcholine, glycolytic metabolites (2,3-diphosphoglycerate, glyceraldehyde phosphate), NO and H2O2 have all been reported to individually increase late INa and [Na+]i in myocytes or myocardium [11, 28, 78, 90–101], although it is not clear whether they act directly on the Na+ channel or indirectly through other targets. Na+ channel blockers (e.g., tetrodotoxin, lidocaine, ranolazine, F15845) have been shown to reduce the rise of Na+ in rat ventricular myocytes and isolated hearts during hypoxia and ischemia, respectively [16, 101–104]. This action of Na+ channel blockers is associated with an improvement of contractile function and reduction of the hypoxia/ischemia-induced increase in the intracellular Ca2+ concentration [91, 93, 101, 103–111].
Late INa is increased in myocytes isolated from failing human and dog hearts [1, 2, 18, 45–47, 51, 112–116]. Inhibition of late INa in myocytes isolated from failing human and dog hearts by ranolazine leads to improvements of relaxation and reductions of afterdepolarizations and variability of duration of the AP [112–114]. These results indicate that late INa is a cause of both Ca2+ overload and reduced repolarization reserve in myocytes from failing hearts.
Late INa was also found to be increased in atrial myocytes isolated from hearts of rabbits with left ventricular hypertrophy. These atrial myocytes had spontaneous EADs and automatic activity [117] that were abolished by tetrodotoxin. Late INa, automaticity, EADs, and delayed afterdepolarizations (DADs) could also be elicited in normal guinea pig atrial myocytes treated with ATX-II or H2O2, and they were abolished by ranolazine [118, 119].
Much progress has been made recently to understand the role of ROS in contributing to an increase of late INa and cardiac Na+/Ca2+ overload. ROS are increased during reoxygenation (reperfusion) following myocardial ischemia [120, 121]. Nitric oxide has been shown to increase late INa in intact neurons and myocytes, and in excised membrane patches from both types of cells [8, 78]. Hydrogen peroxide increases late INa in isolated myocytes and induces arrhythmic activity of myocytes and intact hearts, including EADs, DADs and triggered activity [28, 98]. Arrhythmic activity induced by H2O2 is prevented by ranolazine [98, 119, 122, 123]. An increase of the intracellular Na+ concentration has been shown to cause an NCX-mediated efflux of Ca2+ from mitochondria, reduction of the mitochondrial Ca2+ concentration, and reduction of the mitochondrial NADH/NAD+ ratio [124]. The result is increased formation of ROS by mitochondria [125]. Paradoxically the effect of an increase of [Na+]i is therefore to decrease mitochondrial Ca2+ but increase mitochondrial ROS production, and cytosolic Ca2+.
Elevations of Ca2+/calmodulin [126] and ROS [127–129] can cause activation of CaMKII. CaMKII can phosphorylate NaV1.5, which leads to an increase of late INa [84, 130, 131]. Recently, enhancement of late INa has also been demonstrated to lead to activation of CaMKII; this effect appeared to be dependent on late INa-induced increases of intracellular Na+ and Ca2+ concentrations [132]. CaMKII is upregulated in heart failure [133, 134] and in the infarct border zone [135]. The effects of CaMKII phosphorylations of the Na+ channel to increase late INa, of the L-type Ca2+ channel to increase mode 2 gating, (late ICa) and of ryanodine receptors (RyR2) to increase Ca2+ leak and therefore reduce Ca2+ content of the sarcoplasmic reticulum are all factors contributing to a phenotype of arrhythmogenesis (early- and delayed afterdepolarizations) and contractile dysfunction in heart failure.
Other protein kinases (e.g., the tyrosine kinase Fyn, AMP-activated protein kinase, and protein kinases A and C) in addition to CaMKII are reported to phosphorylate cardiac Na+ channels and increase late INa [59, 60, 136–138]. These kinases play critical roles in signal transduction and “metabolic sensing”. However, their contributions to altered regulation of Na+ channel inactivation and late INa in diseases states is not yet clear.
Electrophysiological Consequences of an Enhanced Late INa
The direct consequence of an enhancement of late INa is an increase of an inward current during the plateau and repolarization (phases 2 and 3) of the cardiac AP, which reduces repolarization reserve. The reduction of repolarization reserve increases the dispersion of repolarization and therefore of AP duration, both locally and across the wall of the heart [139]. An increased dispersion of repolarization creates a substrate for re-entrant activity, and is associated with electrical (T-wave) and mechanical alternans and proarrhythmia, and is predictive of torsade de pointes ventricular tachyarrhythmia [140, 141]. Prolongation of the duration of the AP provides additional time for membrane L-type Ca2+ channels to reactivate, and for the sarcoplasmic reticulum to fill and then release Ca2+ that increases cell membrane NCX (an inward current, ITi), either of which may contribute to an EAD [142]. A failure of Na+ channel inactivation during phase 3 may lead to a larger late INa because the electrochemical driving force for Na+ entry increases as repolarization proceeds. Thus both increased ICa,L and increased INa contribute to EAD formation. EADs commonly occur at low heart rates (with longer AP durations) and can trigger torsade de pointes. The reverse rate dependence of drugs that block hERG K+ current and cause torsade has been demonstrated to be increased by late INa and is reduced by blockers of late INa [143]. Enhancement of late INa may also lead to spontaneous depolarization of the resting membrane potential of a myocyte [119]. Finally, late INa, by its effects to destabilize repolarization and increase the heterogeneity of electrical activity in the myocardium, creates potential paths (substrates) for conduction of re-entrant activity in the heart.
The indirect consequences of an enhancement of late INa are an increase of Na+ influx and of the intracellular Na+ concentration, with subsequent effects on NCX and cellular Ca2+ handling [18]. The effect of an enhanced late INa to increase the concentration of intracellular Na+ will decrease the electrochemical gradient available to extrude Ca2+ via NCX, and thereby contribute to Ca2+ loading of the cytoplasm and therefore of the sarcoplasmic reticulum. When the level of activation of CaMKII is also increased (by high heart rates, catecholamines, an increase of the cytosolic Ca2+ concentration, or ROS-mediated oxidation of the enzyme), CaMKII-mediated phosphorylation of RyR2 (sarcoplasmic reticulum Ca2+ release channels) will occur, and Ca2+ will be released from the sarcoplasmic reticulum during diastole. Phosphorylation of cardiac RyR2 by cAMP-dependent protein kinase A and by CaMKII sensitizes them to Ca2+ and facilitates rapid release of Ca2+ in response to calcium influx (i.e., calcium-induced calcium release) after excitation of the myocyte. However, it also increases the probability of spontaneous Ca2+ release during diastole. The released Ca2+ may exit the cell via NCX. The NCX-coupled exchange of three Na+ for one Ca2+ causes a transient inward current (Iti) and a DAD or EAD [144–146] that may initiate an AP [147] that can trigger arrhythmic activity [148, 149]. The spontaneous “unloading” of Ca2+ from the sarcoplasmic reticulum during diastole also causes a relative depletion of the Ca2+ store available for the following systolic contraction, thus reducing contractility. These events are more likely to occur in the presence of catecholamines and at high heart rates, and in failing hearts, where they lead to Ca2+ and electrical alternans [150, 151].
Late INa and Ca2+ Handling: A Pathological Partnership
As noted above, an increase of late INa causes both an increase of the intracellular Na+ concentration and a prolongation of AP duration, increased activity of NCX in the reverse mode, and increased Ca2+ influx. This is exacerbated during prolonged ischemia, when reductions of cellular ATP and activity of the cell membrane Na+/K+ ATPase allow the intracellular Na+ ([Na+]i) concentration to rise. There is general agreement that the cellular Ca2+ overload that occurs during ischemia and reperfusion is a result of a combination of decreased efflux of Ca2+ ions via the forward mode of NCX and increased influx of Ca2+ ions via the reverse mode of NCX. Direct evidence in support of the participation of reverse mode NCX in intracellular Ca2+ overload during reperfusion or reoxygenation after ischemia is derived from the observations that inhibitors of NCX [152, 153], antisense inhibition of NCX [154], and knockout of NCX [155] markedly decrease either contractile dysfunction or the rise in intracellular Ca2+ in myocardial cells in these settings. For review of NCX in heart failure, see [156].
An increase of the intracellular Ca2+ concentration will cause increased activation of CaMKII. Activation of CaMKII is followed by increased phosphorylation of phospholamban, L-type calcium channels, nitric oxide synthase (to increase formation of NO), RyR2, and Na+ channels. Phosphorylation by CaMKII of both the Na+ channel and the β-subunit of the L-type Ca2+ channel, Cav1.2, increase late INa [84–87], and mode 2 gating of the L-type Ca2+ channel (i.e., late ICa [157]), respectively. Increased formation of NO may also lead to enhancement of late INa [78]. Concomitant CaMKII-mediated increases of late INa and late ICa lead to greater entry of Ca2+ into myocytes but reduction of the Na+ gradient for extrusion of Ca2+ via NCX, thereby facilitating Na+/Ca2+ overload and cardiac dysfunction. The effect of late INa to reduce Ca2+ extrusion via NCX and thereby contribute to Ca2+ overloading and activation of CaMKII, when coupled to the effect of CaMKII to phosphorylate Na+ and Ca2+ channels and increase late INa and late ICa, leads to a pathologic partnership between late INa and activation of CaMKII wherein one begets and amplifies the other.
The Therapeutic Potential of Decreasing Late INa
Selective inhibitors of cardiac late INa have therapeutic potential in the treatment of ischemic heart disease, arrhythmias and heart failure. Inhibitors of late INa are expected to be safe and effective because late INa is enhanced in pathological settings such as ischemia, arrhythmias, and heart failure, but not in healthy myocardium where its inhibition is presumably without consequence. Inhibitors of late INa have been shown to attenuate ionic, metabolic, electrical and contractile dysfunction in preclinical models of hypoxia, ischemia, heart failure, or Na+ overload, suggesting that an increase of late INa plays a critical role in the Na+/Ca2+ overload pathology that underlies ischemia-induced damage to myocardium late INa is augmented. Late INa is augmented, the duration of the AP is prolonged, the dispersion of ventricular repolarization and beat-to-beat variability of AP duration (also referred to as instability) is increased, and EADs are common in ventricular myocytes from dogs and humans with chronic heart failure. The Na+ channel blockers tetrodotoxin, saxitoxin, lidocaine and ranolazine have been shown to shorten the duration and variability of the AP and suppress EADs in ventricular myocytes from failing hearts. For review of the literature regarding the beneficial effects of inhibitors of late INa in models of ischemic heart disease, arrhythmias, and heart failure, see [13, 15–18].
Ranolazine is the most selective inhibitor of late INa in use in current clinical practice [15, 158–162]. It binds to the local anesthetic binding site of the Na+ channel [163] and selectively reduces late relative to peak Na+ current [112, 164]. It does not reduce heart rate, cardiac output, or blood pressure, and is not a systemic vasodilator. Ranolazine does not appear to be arrhythmogenic in spite of its secondary effect to reduce hERG K+ current [162, 165–167], and decreases arrhythmic activity caused by other blockers of this current [168, 169] suggesting that reduction of late INa is sufficient to blunt the pro-arrhythmic actions of hERG blockers.
In summary, Na+ and Na+-induced Ca2+ overloading are characteristic of ischemic and failing hearts and contribute to arrhythmias and a slowing of relaxation (i.e., diastolic stiffness). An increase of late INa is a mechanism for Na+ overloading during hypoxia/reperfusion and heart failure, in patients with gain-of-function SCN5A mutations and possibly in other cardiac diseases. Enhancers of late INa such as ATX-II, H2O2, and ischemic metabolites, and SCN5A mutations cause functional effects that mimic many but not all (e.g., ischemia-induced activation of IK,ATP and shortening of AP duration are not a result of increased late INa) of the effects of ischemia/reperfusion and hypoxia. Selective blockade of late INa reduces mechanical and electrical dysfunction caused by ischemia/reperfusion, arrhythmias, and heart failure, and is thus a rational therapeutic approach to treatment of these diseases.
References
1.
Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–83.PubMed
2.
Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505.PubMed
3.
Wilde AA, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res. 2011;108:884–97.PubMed
4.
Zimmer T, Surber R. SCN5A channelopathies – an update on mutations and mechanisms. Prog Biophys Mol Biol. 2008;98:120–36.PubMed
5.
Amin AS, Asghari-Roodsari A, Tan HL. Cardiac sodium channelopathies. Pflugers Arch. 2010;460:223–37.PubMed
6.
Antzelevitch C, Belardinelli L. The role of sodium channel current in modulating transmural dispersion of repolarization and arrhythmogenesis. J Cardiovasc Electrophysiol. 2006;17 Suppl 1:S79–85.PubMed
7.
George Jr AL. Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–9.PubMed
8.
Hammarstrom AK, Gage PW. Hypoxia and persistent sodium current. Eur Biophys J. 2002;31:323–30.PubMed
9.
Maier LS, Hasenfuss G. Role of [Na+]i and the emerging involvement of the late sodium current in the pathophysiology of cardiovascular disease. Eur Heart J Suppl. 2006;8:A6–9.
10.
Remme CA, Bezzina CR. Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc Ther. 2010;28:287–94.PubMed
11.
Saint DA. The role of the persistent Na(+) current during cardiac ischemia and hypoxia. J Cardiovasc Electrophysiol. 2006;17 Suppl 1:S96–103.PubMed
12.
Saint DA. Persistent (current) in the face of adversity … a new class of cardiac anti-ischaemic compounds on the horizon? Br J Pharmacol. 2009;156:211–3.PubMed
13.
Shryock JC, Belardinelli L. Inhibition of late sodium current to reduce electrical and mechanical dysfunction of ischaemic myocardium. Br J Pharmacol. 2008;153:1128–32.PubMed
14.
Tan HL. Sodium channel variants in heart disease: expanding horizons. J Cardiovasc Electrophysiol. 2006;17 Suppl 1:S151–7.PubMed
15.
Zaza A, Belardinelli L, Shryock JC. Pathophysiology and pharmacology of the cardiac “late sodium current”. Pharmacol Ther. 2008;119:326–39.PubMed
16.
Shryock JC. Role of late sodium channel current in arrhythmogenesis. Card Electrophysiol Clin. 2011;3:125–40.
17.
Saint DA. The cardiac persistent sodium current: an appealing therapeutic target? Br J Pharmacol. 2008;153:1133–42.PubMed
18.
Undrovinas A, Maltsev VA. Late sodium current is a new therapeutic target to improve contractility and rhythm in failing heart. Cardiovasc Hematol Agents Med Chem. 2008;6:348–59.PubMed
19.
Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nature reviews. Cardiology. 2009;6:337–48.PubMed
20.
Liu YM, DeFelice LJ, Mazzanti M. Na channels that remain open throughout the cardiac action potential plateau. Biophys J. 1992;63:654–62.PubMed



