-Adrenoceptors
1.5.2 Mitochondrial Signals
1.5.3 Epicardium
1.6.1 Apela
1.6.3 Pseudo-Noncoding RNAs
1.7.2 Uterus
1.7.3 Blastocyte
1.7.4 Placenta
Oβιoσ βραχυσ, η δετεχνη μακρη, oδεκαιρoσoξυσ, ηδεπειρα σϕαλερη, η δε κρισισ χαλεπη.
[Life is short, art long; opportunity fleeting; experience perilous, and judgment difficult.]
(Hippocrates of Cos [c. 460–c.
460–c. 370], Aphorisms, Sect. I)
370], Aphorisms, Sect. I)
460–c. 370], Aphorisms, Sect. I)In the present handbook, the nouns ailment, disease, illness, malady, pathology, sickness, and syndrome, are often used as synonyms to avoid multiple repetitions of the same word. However, these terms can have distinct meaning.
A syndrome is a collection of signs (observed by individuals external to the patient), symptoms (reported by the patient), and phenomena or features that frequently appear together.1 For example, a metabolic syndrome is not a disease; it underlies a set of diseases and dysfunctions, such as obesity, type-2 diabetes, insulin resistance, dyslipidemia, chronic inflammation, and hypertension, with possible history of stroke and coronary heart disease. A syndrome is, at least originally, less understood than a disease, but can be later explained, or, at least, various pathophysiological aspects involved in the context of a syndrome can be handled (e.g., signaling pathway dysregulation and immunity abnormalities). In other words, the noun usage often continues to be utilized even after an etiology has been successful or when numerous causes that generate the same combination of symptoms and signs have been detected. In fact, some syndromes have a single cause; others have multiple possible causes; the cause of a third category of syndromes remains unknown. Many syndromes are eponymous, i.e., named after the namegiver physicians that first reported the association.
A disease is a disorder in a physiological apparatus or organ that affects the bodily function. It corresponds to any disturbances of the structure–function relation of a tissue, organ, or physiological apparatus manifested by a characteristic set of symptoms and signs and whose etiology and prognosis can be usually identified and predicted. A disease is a condition characterized usually by at least two criteria among three basic elements: (1) recognized etiologic agent (cause); (2) identifiable group of signs and symptoms; and (3) consistent anatomopathologic alterations. Some diseases can cause a particular syndrome. The term disease is more concrete than illness, which can also include mental aspects.
An illness is an abnormal process in which aspects of the social, physical, emotional, and/or intellectual function of a person are impaired compared with previous behavior. It is a malady of either body or mind, the symptoms of which may be physically unobservable. Illness encompasses the subjective characteristics of the disturbance as a whole rather than only objective symptoms.
A sickness commonly results from difficulty of adjusting to environmental conditions (e.g., altitude, sea, car, decompression, and radiation sickness).
Cardiovascular disease refers to any dysfunctioning or lesion that affects the cardiovascular apparatus, the cardiac pump, as well as the irrigation and drainage circuits, mainly those of the heart, brain, and kidneys.
1.1 Gene Transcription in Cardiovascular Diseases
The transcriptional control of the cardiovascular function involves programs of gene activation and suppression. Transcription factors, chromatin remodelers, and histone modifiers, that is, a set of coactivators and corepressors cooperates to influence chromatin state and hence controls accessibility of nucleosomal DNA, as well as directly regulates gene expression in the heart.
During cardiogenesis, chamber specification is controlled by temporal and spatial expression of cardiac transcription factors such as heart and neural crest derivatives expressed factor HAND, GATA-binding protein GATA4, myocyte enhancer factor MEF2, cardiac-specific NK2 homeobox-encoded factor NKx2-5, and T-box factor TBx5 [1]. Transcription factors GATA4, MEF2c, MEF2d, and myogenic differentiation factor MyoD, are involved in transcriptional programs launched by signals that control pathological gene expression in the heart [1].
1.1.1 Chromatin Remodelers
Chromatin architecture and its modification is involved in gene regulation as well as DNA repair, recombination, and replication. In the cell nucleus, DNA is packaged by histones to form structured nucleosome units. Chromatin remodeling is governed by two categories of enzymes: histone modifiers and ATP-driven nucleosome repositioners.
Transcriptional controllers encompass:
Catalyzers of chemical modifications (i.e., acetylation and methylation) of histone proteins within the chromatin by histone acetyltransferases (HATs), deacetylases (HDAC), and methyltransferases (HMT);
Conditioners of the chromatin structure and topology such as chromatin remodelers and helicases of the switch/sucrose nonfermentable(Swi/SNF) ATPase complex implicated in cardiogenesis and heart diseases, in particular, Swi/SNF-related, matrix-associated, actin-dependent regulator of chromatin SMARCa2 and SMARCa4;2 and
Noncoding RNAs (e.g., Braveheart) that interact with chromatin.
HDACs and HMTs are associated with certain cardiac pathologies (Tables 1.1 and 1.2). Mutations in genes encoding HMTs may contribute to congenital heart disease.
Table 1.1
Chromatin modifications and histone deacetylase (HDAC)-dependent pathological gene expression in the heart. (Source: [1])
Type | Disease | Target genes |
|---|---|---|
HDAC1 | Cardiac hypertrophy | Ncx1 BECN1 (autophagy related beclin-1) |
HDAC2 | Cardiac hypertrophy, fibrosis | Anp, Bnp Klf, Yy1 |
Hsp70, ATG5, BECN1 | Inpp5f, Gsk3β | |
HDAC3 | Cardiac hypertrophy, fibrosis | Tnfsf1 |
HDAC4 | Heart failure, ischemia | SUV39H1 HIF1A |
HDAC5 | Cardiac hypertrophy Ischemia | NKX2.5, p300 Ncx1 |
HDAC6 | Cardiac hypertrophy (induced by angiotensin-2; pro) | |
HDAC9 | Cardiac hypertrophy | FOXP3 |
Table 1.2
Chromatin modifications associated with histone methylation and demethylation in cardiac diseases. The MYH6 and MYH7 genes encode α- and β-cardiac myosin heavy chain, respectively. (Source: [1])
Type | Function | Pathology (role) | Target gene |
|---|---|---|---|
ASxL2 | H3K 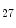 me me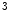 | Ischemic and idiopathic DCM | MYH6/7 |
KMT1c | H3K 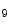 me me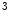 | Ischemia (pro) | Sirt1 |
KMT3c | H3K 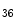 me me | ND | ND |
KMT3d | H3K 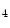 me me | Arrhythmia (pro) | ND |
KMT4 | H3K 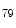 me me | DCM (anti) | DMD (dystrophin) |
KMT6 | H3K 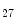 me me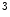 | ND | Anp, Bnp, MYH6/7 |
KDM3a | ND | ND | ND |
KDM4a | H3K 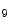 demethylase demethylase | Cardiac hypertrophy, HCM, ICM (pro) | Anp Fhl1 |
KDM4b | H3K 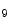 demethylase demethylase | DCM, ICM (pro) | Anp, Bnp |
KDM6a | H3K 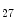 demethylase demethylase | Hypoxia (pro) | Nos3 |
KDM6b | H3K 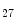 demethylase demethylase | Hypoxia (pro) | Nos3 |
PaxIP1 | Cofactor of KMT2c/d (H3K  me me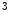 ) ) | Ventricular arrhythmia (anti) | KCNIP2 |
HDM | H3K 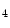 me me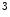 | Heart failure | DUX4 |
H3K 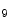 me me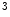 | |||
H3K 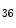 me me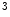 |
ASxL additional sex combs-like protein, HDM histone demethylase, KDM histone Lys (K)-specific demethylase, kKMT histone Lys (K) 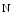 methyltransferase, PaxIP paired box (Pax)-interacting with transcription-activation domain protein, anti impedes disease, pro favors disease, DCM dilated cardiomyopathy, HCM hypertrophic cardiomyopathy, ICM ischemic cardiomyopathy, DUX double homeobox gene, FHL four and a half LIM domain-containing protein, ND not described
methyltransferase, PaxIP paired box (Pax)-interacting with transcription-activation domain protein, anti impedes disease, pro favors disease, DCM dilated cardiomyopathy, HCM hypertrophic cardiomyopathy, ICM ischemic cardiomyopathy, DUX double homeobox gene, FHL four and a half LIM domain-containing protein, ND not described
methyltransferase, PaxIP paired box (Pax)-interacting with transcription-activation domain protein, anti impedes disease, pro favors disease, DCM dilated cardiomyopathy, HCM hypertrophic cardiomyopathy, ICM ischemic cardiomyopathy, DUX double homeobox gene, FHL four and a half LIM domain-containing protein, ND not describedClass-1 nuclear histone deacetylases (HDAC1–HDAC3 and HDAC8), class-2 HDACs, which shuttle between the cytosol and nucleus and can further be subdivided into subclass-2a (HDAC4, HDAC5, HDAC7, and HDAC9) and -2b (HDAC6 and HDAC10), and class-4 HDACs (HDAC11), which localize to the nucleus and cytosol and depend on zinc. Class-2a HDACs repress cardiac hypertrophy. On the other hand, class-3 HDACs, or sirtuins (SIRT1–SIRT7), rely on nicotinamide adenine dinucleotide (NAD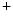 ). They reside in both nucleus and cytoplasm.
). They reside in both nucleus and cytoplasm.
). They reside in both nucleus and cytoplasm.The class-2b cytoplasmic histone deacetylase HDAC6, which lodges in striated (cardiac and skeletal) myocytes and deacetylates cytoskeletal proteins, is involved in adverse remodeling induced by chronically signaling angiotensin-2 [2]. HDAC6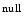 mice develop cardiac hypertrophy and fibrosis, but without cardiac function alteration, in response to angiotensin-2 administration during up to 8 weeks, whereas wild-type mice develop systolic dysfunction. Inhibition of HDAC6 preserves systolic function.
mice develop cardiac hypertrophy and fibrosis, but without cardiac function alteration, in response to angiotensin-2 administration during up to 8 weeks, whereas wild-type mice develop systolic dysfunction. Inhibition of HDAC6 preserves systolic function.
mice develop cardiac hypertrophy and fibrosis, but without cardiac function alteration, in response to angiotensin-2 administration during up to 8 weeks, whereas wild-type mice develop systolic dysfunction. Inhibition of HDAC6 preserves systolic function.Histone acetylation and methylation that result from developmental programs of the heart dictate cardiac gene expression. Histone Lys 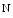 methyltransferase KMT63 and paired box (Pax) transactivation domain-interacting protein PaxIP1 (or PTIP) are two HMTs involved in chromatin condensation. The former participates in cardiac myosin heavy chain (MHC) gene regulation, as it interacts with a long noncoding RNA in chromatin [1].
methyltransferase KMT63 and paired box (Pax) transactivation domain-interacting protein PaxIP1 (or PTIP) are two HMTs involved in chromatin condensation. The former participates in cardiac myosin heavy chain (MHC) gene regulation, as it interacts with a long noncoding RNA in chromatin [1].
methyltransferase KMT63 and paired box (Pax) transactivation domain-interacting protein PaxIP1 (or PTIP) are two HMTs involved in chromatin condensation. The former participates in cardiac myosin heavy chain (MHC) gene regulation, as it interacts with a long noncoding RNA in chromatin [1].Trimethylation H3K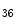 me
me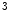 is typically associated with transcriptional elongation and DNA methylation that cooperate to regulate the expression of double homeobox Dux4 gene in the failing human heart [1].
is typically associated with transcriptional elongation and DNA methylation that cooperate to regulate the expression of double homeobox Dux4 gene in the failing human heart [1].
me is typically associated with transcriptional elongation and DNA methylation that cooperate to regulate the expression of double homeobox Dux4 gene in the failing human heart [1].Histone-3 trimethylation at Lys4 (e.g., H3K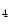 me
me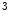 ) and some types of acetylation (H3K
) and some types of acetylation (H3K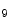 ac and H4K
ac and H4K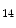 ac) are generally correlated with permissive gene transcription, as they weaken the connection of histone proteins and DNA.
ac) are generally correlated with permissive gene transcription, as they weaken the connection of histone proteins and DNA.
me) and some types of acetylation (H3Kac and H4Kac) are generally correlated with permissive gene transcription, as they weaken the connection of histone proteins and DNA.In particular, H3K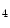 me
me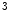 enables the expression of the KCNIP2 gene that encodes K
enables the expression of the KCNIP2 gene that encodes K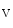 channel-interacting protein KChIP2 involved in repolarization [1]. Expression of the KCNIP2 gene associated with reduced H3K
channel-interacting protein KChIP2 involved in repolarization [1]. Expression of the KCNIP2 gene associated with reduced H3K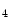 me
me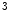 at its promoter falls in heart failure. cAMP-responsive element-binding protein (CREB)-binding protein (CBP) and P300 are two HATs detected on activated genes in the left and right ventricles of the heart. Binding of cardiac transcription factors, such as SRF and GATA4, may be mediated at least partly by P300 acetyltransferase [1].
at its promoter falls in heart failure. cAMP-responsive element-binding protein (CREB)-binding protein (CBP) and P300 are two HATs detected on activated genes in the left and right ventricles of the heart. Binding of cardiac transcription factors, such as SRF and GATA4, may be mediated at least partly by P300 acetyltransferase [1].
me enables the expression of the KCNIP2 gene that encodes K channel-interacting protein KChIP2 involved in repolarization [1]. Expression of the KCNIP2 gene associated with reduced H3Kme at its promoter falls in heart failure. cAMP-responsive element-binding protein (CREB)-binding protein (CBP) and P300 are two HATs detected on activated genes in the left and right ventricles of the heart. Binding of cardiac transcription factors, such as SRF and GATA4, may be mediated at least partly by P300 acetyltransferase [1].The histone modifiers , additional sex comb-like protein ASxL2 and histone Lys 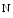 methyltransferase KMT6, interact at MHC promoters [1].
methyltransferase KMT6, interact at MHC promoters [1].
methyltransferase KMT6, interact at MHC promoters [1].On the other hand, methylation of H3K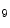 and H3K
and H3K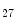 me
me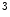 and deacetylation are correlated with gene transcription suppression, as they cause chromatin condensation (i.e., transcriptionally repressive chromatin conformation) [1]. In mouse models, ischemic reperfusion provokes H3K
and deacetylation are correlated with gene transcription suppression, as they cause chromatin condensation (i.e., transcriptionally repressive chromatin conformation) [1]. In mouse models, ischemic reperfusion provokes H3K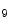 me
me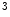 at the Sirt1 promoter, hence suppressing the Sirt1 gene transcription.
at the Sirt1 promoter, hence suppressing the Sirt1 gene transcription.
and H3Kme and deacetylation are correlated with gene transcription suppression, as they cause chromatin condensation (i.e., transcriptionally repressive chromatin conformation) [1]. In mouse models, ischemic reperfusion provokes H3Kme at the Sirt1 promoter, hence suppressing the Sirt1 gene transcription.However, whereas deacetylation of histone tails is linked to transcriptional suppression, deacetylation of nonhistone proteins often activates gene expression.
The Polycomb repressive complexes PRC1 and PRC2 cause gene silencing via intrinsic HMT activity. The PRC2 holoenzyme is a complex of HMTs, which leads to H3K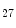 me
me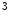 modification.
modification.
me modification.The regulation of gene expression involves the coordinated actions of the transcriptional machinery and regulators (i.e., activators or repressors at gene promoters). Gene transcription can result from a loss of inhibitory methylation and a gain of stimulatory acetylation in a given Lys residue (e.g., K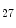 ) of histone H3.
) of histone H3.
) of histone H3.1.1.2 Gene Transcription and Mechanical Factors
At a given station, the velocity and possibly the direction of blood flow change (e.g., in limb arteries, but not in cerebral arteries) during the cardiac cycle, in addition to spatial variations along the vascular circuit. The endothelium is sensitive to hemodynamic shear stresses applied at its luminal surface (Vol. 5, Chap. 9. Vascular Endothelium).
Blood flow via hemodynamic stress exerted on the wall-wetted surface as well as within the wall can promote expression of genes that encode antiatherogenic, antithrombotic, and anti-inflammatory factors [3].
1.1.2.1 Sheared Endothelium
The vascular endothelium is a thin monolayer of cells that line the luminal surface of all blood and lymph vessels. This barrier controls the exchange of water, electrolytes, macromolecules, and cells between the vascular lumen and surrounding tissue. It regulates leukocyte adhesion and transendothelial migration, as well as platelet aggregation via the expression of adhesion molecules. It supervises smooth myocyte function via vasoactive substances (e.g., nitric oxide, prostacyclin, and endothelin-1).
Inside the vascular wall, cyclic strain and stretch result form the normal hemodynamic stress, blood pressure. The latter indeed cyclically generates axial and circumferential tension applied to both intimal endothelial and medial smooth muscle cells. Shear is the mechanical force created by blood particles decelerated (slowed) by the immobile solid wall or by adjacent slower blood particle (in the local normal direction to wall surface) on its wall-facing surface and accelerated (dragged, i.e., gaining momentum) by adjacent quicker blood particle. At the wetted endothelial surface, the moving blood particle exerts a tangential force and cyclically shears the luminal border of endotheliocytes.
Shear stress sensors of endotheliocytes include ion channels, plasmalemmal receptors (GPCRs and RTKs), adhesion molecules connected to the cytoskeleton and matrix fibers, and caveolae, as well as the glycocalyx and primary cilia. Mechanoreceptors convert mechanical cues into chemical signals that control cell function and fate via stored molecules and regulation of gene expression.
Cultures of endotheliocytes in the absence of mechanical stress do not mimic the physiological condition. Once endotheliocytes are exposed to shear stress (the common situation in vivo), expression of some genes increases with respect to that at rest (unphysiological condition), such as those that encode NOS3, thrombomodulin, cytochrome-P450, heme oxygenase-1, diaphorase-4, PECAM1, regulators of G-protein, and KLF2 and KLF4 [4]. On the other hand, the expression of genes encoding inflammatory and thrombogenic agents (e.g., VCAM1, E-selectin, TGFβ, and BMP4) is downregulated.
Transcription factors of the Krüppel-like factor family regulate responses to shear stress and engender anti-inflammatory and anticoagulant transcripts. The Krüppel-like factor KLF2 elicits expression of atheroprotective genes, such as those encoding NOS3, thrombomodulin, MAPK9, and von Willebrand factor, and impedes transcription of proatherogenic genes, such as those encoding NFκ B, BMP4, interleukin-8 (CXCL8), and Serpin-E1) [4]. However, endothelial signature genes, such as those encoding VEGFR1 and VEGFR2, TIE1 and TIE2, PDGF, VEGF, and cadherin-5 are not significantly influenced by KLF2. KLF2 and KLF4 are regulated via the MAP2K5–ERK5 pathway.
1.1.2.2 Hemodynamic Stress and Vascular Remodeling
Hemodynamic stress field on and in the vascular mural domain influences not only the vascular tone, that is, the local size of the vascular lumen and, hence, the flow resistance, but also outward and inward vascular remodeling (Vol. 5, Chap. 11. Tissue Development, Repair, and Remodeling). Atherosusceptibility refers to an adaptive chronic low-level inflammatory state that ensures the endothelial function with an increased likelihood of atherogenesis. Atherosclerotic plaques do not distribute evenly over the arterial bed, but localize to predilection sites with low-magnitude velocity at bifurcations, side branches, and some regions of curved arteries, where a reduced blood flow velocity supports uptake and activation of molecules and inflammatory cells. In addition, some shear stress patterns provoke specific plaque compositions in a high-cholesterol environment, a low and oscillatory shear stress favoring plaque stabilization with respect to a low steady regime [4].
1.1.2.3 Mechanical Stress and Stem Cells
The adult heart is not a fully terminally differentiated organ, as a cardiomyocyte turnover exists with a rate estimated at about 1 %/year in young adults and 0.5 %/year in the elderly.
Mechanical stresses influence vascular injury and repair. In particular, blood flow impacts maturation of circulating or resident stem cells into functional vascular cells. A heterogeneous population of stem and progenitor cells may exist in different layers of the vessel wall.
In healthy walls, stem cells are quiescent and localize to parietal stem cell niches. SCFR+, Sca1+, Lin- or Sox17+, Sox10+, and S100β+ multipotent stem cells can differentiate into many cell types. HCAM+, NT5E+, Thy1+, CD34-, and PTPRc- mesenchymal stem cells constitute a second stem cell population. The third population is formed by CD34+, NG2+, and PDGFRβ+ pericytes [5].
Vascular stem cells can differentiate into endotheliocytes or migrate across the vessel wall and subsequently differentiate into intimal smooth myocytes [5].
Endothelial progenitor cells reside within the endothelium as well as in the intimal subendothelial layer, where they can form neovessels, although they are normally dormant [5].
Pericyte progenitor cells lodge around adventitial vasa vasorum. They support angiogenesis [5].
In the blood vessel wall, multipotent vascular stem cells can differentiate into neurons and mesenchymal stem cell-like cells that subsequently differentiate into smooth myocytes [5]. At least some lesional smooth myocytes with a proliferative and synthetic phenotype derive from these stem cells rather than smooth myocytes that dedifferentiated and migrated from the media.
Resident stem cells may be released into the blood circulation. They can differentiate into vascular cell lineage to repair damaged vessels or form new microvessels in damaged tissues. In addition, bone marrow stem cells can generate circulating endothelial progenitor cells. Smooth myocyte progenitors also flow in blood and can contribute to the pathogenesis of vascular diseases [5].
Vessel-resident (intimal, medial, and adventitial) and bloodborne vascular progenitor cells differentiate according to their location within the vascular wall into smooth myocytes when stretch is the main actor and endotheliocytes when shear is the major player, despite its lower magnitude, as the shear sensor density may be higher than that of stretch transducers [5].
1.1.3 Gene Transcription in Cardiomyocytes and Glucocorticoids
The stress response relies on the hypothalamic–pituitary–adrenal axis. This activated axis triggers the release of glucocorticoids from the adrenal gland. Glucocorticoids have potent anti-inflammatory and immunosuppressive effects. These stress hormones connect to the glucocorticoid receptor (GR), a member of the nuclear receptor superfamily of ligand-dependent transcription factors (NR3c1).
Expression of genes involved in GR-deficient hearts include [6]: (1) the maintenance of cardiac contractility (DMD [dystrophin] and RYR2 [cardiac ryanodine receptor]); (2) repression of adverse cardiac hypertrophy (Klf15 [Krüppel-like factor-15 and TRIM63 [Tripartite motif-containing protein-63; also known as Muscle-specific RING finger ubiquitin–protein ligase (MURF1)]); (3) promotion of cardiomyocyte survival (Ptgds [prostaglandin-D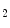 synthase]); and (4) inhibition of inflammation decreases (LCN2 [lipocalin-2], and Zfp36 [Zinc finger protein-36 or tristetraprolin]). The dysregulated glucocorticoid-responsive target genes (lowered expression) encode many transcription factors, plasmalemmal receptors, and effectors of G protein-coupled receptor (GPCR) and mitogen-activated protein kinase (MAPK) signaling [6]. A glucocorticoid signaling deficiency to cardiomyocyte thus causes maladaptive cardiac hypertrophy, heart failure, and death [6].
synthase]); and (4) inhibition of inflammation decreases (LCN2 [lipocalin-2], and Zfp36 [Zinc finger protein-36 or tristetraprolin]). The dysregulated glucocorticoid-responsive target genes (lowered expression) encode many transcription factors, plasmalemmal receptors, and effectors of G protein-coupled receptor (GPCR) and mitogen-activated protein kinase (MAPK) signaling [6]. A glucocorticoid signaling deficiency to cardiomyocyte thus causes maladaptive cardiac hypertrophy, heart failure, and death [6].
synthase]); and (4) inhibition of inflammation decreases (LCN2 [lipocalin-2], and Zfp36 [Zinc finger protein-36 or tristetraprolin]). The dysregulated glucocorticoid-responsive target genes (lowered expression) encode many transcription factors, plasmalemmal receptors, and effectors of G protein-coupled receptor (GPCR) and mitogen-activated protein kinase (MAPK) signaling [6]. A glucocorticoid signaling deficiency to cardiomyocyte thus causes maladaptive cardiac hypertrophy, heart failure, and death [6].In addition to GR, cardiomyocytes express the mineralocorticoid receptor (MR; NR3c2) that binds aldosterone and glucocorticoids with similar high affinity. Glucocorticoids typically circulate at much higher levels than aldosterone, and cardiac-specific MR may be primarily occupied by glucocorticoids [6]. However, the contribution of MR is negligible.
Patients who bear a single nucleotide polymorphism (A3669G) in the GR gene are characterized by an increased expression of the dominant-negative receptor variant GRβ and concomitant glucocorticoid resistance. They have an augmented risk of coronary artery disease, adverse cardiac hypertrophy, systolic dysfunction, and heart failure [6]. In addition, patients with Addison’s disease identified by a decreased production of glucocorticoids have heart failure.
1.2 Myocardial Regeneration Capacity
Regeneration of the cardiac wall requires the recreation of the myocardial structure with its stroma, nodal tissue, and vasculature and restoration of the contractile function of quasi-synchronized cardiomyocytes due to distinct electrophysiological features according to regional and intramural localization. The cardiac regeneration may be achieved by stimulating the proliferation of cardiac progenitors as well as cardiomyocytes.
Whereas the mammalian fetal or neonatal heart can repair and generate a new contractile myocardium after damage, after a later myocardial loss, fibrosis predominates. The capacity of the heart to repair after damage remain thus restricted.
1.2.1 Cardiomyocytes Before and After Birth
The heart is the first organ built during embryo- and fetogenesis. During the fetal life, differentiated cardiomyocytes enter the cell cycle. The primitive heart formed in the early stages of cardiac specification mostly consists of quiescent cells. As development progresses, cardiomyocytes at the outer curvature resume proliferation to form the future chambers. On the other hand, cardiomyocytes at the inner curvature do not enter the cell cycle and will form the conduction system.
Embryonic and neonatal cardiomyocytes are capable of synthesizing, assembling, and disassembling their contractile proteins via the assembly of contractile structures for chromosome segregation and cytokinesis when they proliferate [7].
Proliferation and differentiation are not mutually exclusive in the developing as well as adult heart in some animal species that can regenerate their cardiac pump. For example, the damaged zebrafish heart reactivates a developmental program involving the expansion of GATA4+ cardiomyocytes to achieve regeneration [7].
However, the cardiomyocyte proliferation is limited in mammals. In humans, the cardiomyocyte stops dividing early after birth, possibly due to intracellular activation of the cyclin-dependent kinase inhibitors CKI1b and CKI2c, when breathing brings a sudden and marked increase in oxygenation ( the arterial oxygen partial pressure 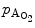 ranges from 2.4 to 3.7 kPa [18–28 mmHg] which, during the fetal life, rises to 13.3 kPa [100 mmHg]) [7]. At birth, oxygen free radicals and reactive oxygen species (ROS) may stop cardiomyocytes proliferation and favor their terminal differentiation.
ranges from 2.4 to 3.7 kPa [18–28 mmHg] which, during the fetal life, rises to 13.3 kPa [100 mmHg]) [7]. At birth, oxygen free radicals and reactive oxygen species (ROS) may stop cardiomyocytes proliferation and favor their terminal differentiation.
ranges from 2.4 to 3.7 kPa [18–28 mmHg] which, during the fetal life, rises to 13.3 kPa [100 mmHg]) [7]. At birth, oxygen free radicals and reactive oxygen species (ROS) may stop cardiomyocytes proliferation and favor their terminal differentiation. The neonatal cardiomyocytes lack a developed T-tubular network (microscopic invaginated segments of the sarcolemma); the myofibrils are located near the sarcolemma. In growing cardiomyocytes, T-tubules develop within a few days after birth. Caveolin-3 localized to the nanoscopic invaginations of the sarcolemma, caveolae, transiently associates with primitive T-tubules. The location of various K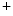 , Na
, Na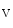 1, and Ca
1, and Ca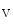 1.2 channels relies on caveolae.
1.2 channels relies on caveolae.
, Na1, and Ca1.2 channels relies on caveolae. Bridging integrator BIn1 (or 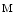 amphiphysin-2) is a major agent of T-tubule genesis and transfer of Ca
amphiphysin-2) is a major agent of T-tubule genesis and transfer of Ca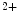 handling proteins in T-tubules. This membrane scaffold initiates T-tubule genesis in striated myocytes, asit induces membrane curvature. In addition, it determines the delivery of microtubule-transported membrane proteins. In particular, it forms BIn1–Ca
handling proteins in T-tubules. This membrane scaffold initiates T-tubule genesis in striated myocytes, asit induces membrane curvature. In addition, it determines the delivery of microtubule-transported membrane proteins. In particular, it forms BIn1–Ca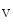 1.2 clusters and tethers microtubules to membrane, thereby supporting Ca
1.2 clusters and tethers microtubules to membrane, thereby supporting Ca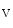 1.2 delivery to the T-tubular membrane [8]. It especially anchors microtubules and Ca
1.2 delivery to the T-tubular membrane [8]. It especially anchors microtubules and Ca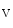 1.2 at junctional membrane complexes. These specialized nanodomains connect ion channel clusters between transverse tubule and sarcoplasmic reticulum that are involved in excitation–contraction coupling that links membrane depolarization to mechanical contraction. A small entry of extracellular Ca
1.2 at junctional membrane complexes. These specialized nanodomains connect ion channel clusters between transverse tubule and sarcoplasmic reticulum that are involved in excitation–contraction coupling that links membrane depolarization to mechanical contraction. A small entry of extracellular Ca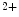 through Ca
through Ca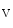 1.2a causes a large Ca
1.2a causes a large Ca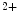 release from its major intracellular store through RyR2 channels. This Ca
release from its major intracellular store through RyR2 channels. This Ca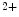 -induced Ca
-induced Ca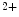 release (CICR) inactivates Ca
release (CICR) inactivates Ca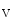 1.2a for repolarization. Altered splicing and mutations of Bin1 are involved in myopathies.
1.2a for repolarization. Altered splicing and mutations of Bin1 are involved in myopathies.
amphiphysin-2) is a major agent of T-tubule genesis and transfer of Ca handling proteins in T-tubules. This membrane scaffold initiates T-tubule genesis in striated myocytes, asit induces membrane curvature. In addition, it determines the delivery of microtubule-transported membrane proteins. In particular, it forms BIn1–Ca1.2 clusters and tethers microtubules to membrane, thereby supporting Ca1.2 delivery to the T-tubular membrane [8]. It especially anchors microtubules and Ca1.2 at junctional membrane complexes. These specialized nanodomains connect ion channel clusters between transverse tubule and sarcoplasmic reticulum that are involved in excitation–contraction coupling that links membrane depolarization to mechanical contraction. A small entry of extracellular Ca through Ca1.2a causes a large Ca release from its major intracellular store through RyR2 channels. This Ca-induced Ca release (CICR) inactivates Ca1.2a for repolarization. Altered splicing and mutations of Bin1 are involved in myopathies. In cardiomyocytes, junctophilin-2 encoded by the JPH2 gene is a major structural protein in T-tubules that forms junctional membrane complexes. Junctophilin-2 spans the junctional distance (∼12 nm cleft) and tethers the plasma membrane to that of the sarcoplasmic reticulum to facilitate Ca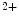 handling [9]. Its N-terminus is attached to the T-tubular sarcolemma and its C-terminal transmembrane domain is embedded in the sarcoplasmic reticulum. Junctophilin-2 is necessary for the development of postnatal T-tubules and stabilization of junctional membrane complexes in mature cardiomyocytes. Junctophilin-2 also interacts with caveolin-3 [9]. Its deficiency by mutations or expression downregulation is linked to hypertrophic cardiomyopathy, arrhythmias, and heart failure.
handling [9]. Its N-terminus is attached to the T-tubular sarcolemma and its C-terminal transmembrane domain is embedded in the sarcoplasmic reticulum. Junctophilin-2 is necessary for the development of postnatal T-tubules and stabilization of junctional membrane complexes in mature cardiomyocytes. Junctophilin-2 also interacts with caveolin-3 [9]. Its deficiency by mutations or expression downregulation is linked to hypertrophic cardiomyopathy, arrhythmias, and heart failure.
handling [9]. Its N-terminus is attached to the T-tubular sarcolemma and its C-terminal transmembrane domain is embedded in the sarcoplasmic reticulum. Junctophilin-2 is necessary for the development of postnatal T-tubules and stabilization of junctional membrane complexes in mature cardiomyocytes. Junctophilin-2 also interacts with caveolin-3 [9]. Its deficiency by mutations or expression downregulation is linked to hypertrophic cardiomyopathy, arrhythmias, and heart failure.1.2.2 Oxygen Concentration and Cardiomyocyte Proliferation
The epicardium, which can be considered as a cardiac stem cell niche, is subjected to a relative hypoxia that yet enables the proliferation of cardiac progenitor cells. Epicardial progenitor cells are activated during embryogenesis and generate cardiomyocytes, endotheliocytes, and vascular smooth myocytes, as well as in adulthood after injury, once they are stimulated by thymosin-β4 [7]. The latter activates epicardial progenitors by upregulating antioxidative enzymes, thereby attenuating oxidative stress in these cells. In addition, a low oxygen partial pressure promotes the proliferation of human cardiomyocyte progenitors [7].
1.2.3 Mechanical Load and Cardiomyocyte Proliferation
In addition, cardiomyocytes are subjected to an increased mechanical load after birth. Cells of the inner wall surface, which sense shear in addition to stretch, are the first to exit cell cycle [7]. Cardiomyocytes are equipped with stress and strain sensors in Z disks and titin filaments that can modify the transcriptional activity. Stretch can provoke the production of growth factors and cytokines, such as IL6 and IGF1, which serve as paracrine controllers of cardiomyocyte proliferation [7].4 Mechanical strain increases the intracellular level of ROS that promote cardiomyocyte differentiation and block their division.
1.2.4 Growth Factors and Cardiomyocyte Proliferation
Neighboring fibroblasts, endotheliocytes, smooth myocytes, adipocytes, and inflammatory cells influence cardiomyocyte proliferation, as they release paracrine messengers, in particular growth factors and cytokines.
In neonatal cardiomyocytes, fibroblast growth factor FGF2 promotes DNA synthesis via activated protein kinase-C [7]. In the adult myocardium, the FGF2 activity is potently inhibited by FGF16, which is preferentially expressed in the postnatal heart.
In neonatal cardiomyocyte cultures, platelet-derived growth factor supports cell proliferation, as it represses the G1-phase inhibitor CKI1b, activates PKB, and inhibits GSK3β [7].
Inflammatory meukocytes secrete the TNFSF12 cytokine that helps cardiomyocyte proliferation upon binding to TNFRSF12a receptor expressed by neonatal cardiomyocytes and downregulated in adulthood, except in pathological conditions [7].
Interleukin-6, which is abundantly secreted in the heart by both epicardiocytes and hypoxic adipose stromal cells, augments the proliferation rate of cultured slow-dividing neonatal cardiomyocytes [7].
Neuregulin-1, a member of the EGF family, is an inducer of adult cardiomyocyte proliferation via HER4 receptor [7]. Neuregulin-1 is first expressed by the endocardial endothelium of neonatal hearts, where it participates in trabeculation and cushion formation, and later by endotheliocytes. Periostin is another secreted molecule that may increase proliferation of both neonatal and adult cardiomyocytes via integrins and the PI3K pathway [7].
1.2.5 Extracellular Matrix and Cardiomyocyte Proliferation
The composition of the extracellular matrix contributes to the control of the rate of cardiomyocyte proliferation before and after birth. The cardiomyocyte division in the developing heart correlates with the regulated synthesis of matrix proteic constituents and integrins. In the fetal myocardium, the extracellular matrix that is enriched in fibronectin supports cardiomyocyte proliferation [7].
On the other hand, an elevated concentration of collagen-1 impedes cardiomyocyte proliferation after birth [7]. Focal adhesion kinase (FAK) that assists fibronectin binding to integrins facilitates cardiomyocyte proliferation during cardiogenesis heart development. FAK-related nonkinase (FRNK), the noncatalytic FAK C-terminus, is transiently expressed in the postnatal heart with peak levels occurring just prior to the withdrawal of cardiomyocytes from the cell cycle [7]. Therefore, FRNK may dampen FAK-dependent cardiomyocyte cell cycle progression and facilitate cell cycle exit in the postnatal myocardium.
1.2.6 MicroRNAs and Cardiomyocyte Proliferation
Cardiogenesis as well as cardiac homeostasis in adulthood is controlled by several different microRNAs [7]. These genome-encoded small RNAs mediate posttranscriptional silencing, thereby preventing unwanted gene expression.
The expression of microRNA-195, a member of the MIR15 family, is induced when cardiomyocytes exit the cell cycle. MiR29a and miR133 also suppress cardiomyocyte proliferation.
On the other hand, miR199a-3p and miR590-3p are able to cause cell cycle reentry and cytokinesis of cultured adult cardiomyocytes [7]. In vivo, these miRs specifically promoted cardiomyocyte (but not fibroblast) proliferation in both neonatal and adult hearts after myocardial infarction.
MicroRNA-499 promotes neonatal cardiomyocyte proliferation via Sox6 and cyclin-D1 [7]. The microRNA-17–92 cluster is also involved in cardiomyocyte proliferation, as its overexpression engenders cardiomyocyte proliferation in embryonic, postnatal, and adult hearts.
1.2.7 Genetic Manipulations and Cardiomyocyte Proliferation
The cardiomyocyte proliferation can be stimulated by different types of extracellular molecules. In the mouse adult heart, a slow-rate cardiomyocyte renewal is sustained by the division of preformed cardiomyocytes [7].
The exogenous administration of selected growth factors is capable of inducing neonatal and, in some instances, also adult cardiomyocyte proliferation [7]. The cardiac regeneration in the adult heart may also be achieved by reactivating the cardiogenesis program.
In addition, certain diffusible factors can regulate the proliferation and cardiac commitment of endogenous or implanted stem cells. The type of cardiomyocytes (nodal, atrial, or ventricular) can be identified by electrophysiological properties (e.g., shape of action potential waveform and duration and amplitude of generated action potentials). The characterization of action potentials enables automated categorization of cardiomyocyte populations from embryoid bodies (i.e., aggregates of pluripotent stem cells formed from embryonic or induced pluripotent stem cells by reprogramming somatic cells) for optimal implantation [10]. The Notch pathway operates between adjacent cells expressing Notch ligands (Jag1 and Jag2 and DLL1 and DLL3–DLL4) and receptors (Notch-1–Notch-4; Vol. 3, Chap. 10. Morphogen Receptors). In the developing heart, Notch modulates cardiomyocyte survival, cardiac stem cell differentiation, and angiogenesis and regulates trabeculation, cardiomyocyte proliferation, and valve formation [11]. In the postnatal heart, the Notch axis controls cardiac precursor expansion and differentiation, thereby sustaining cardiomyocyte proliferation.
However, in adult mice hearts, reactivation of the Notch pathway does not assist cardiac regeneration after myocardial infarction [7]. Therefore, at least in adult rodent hearts, cardiac regeneration does not necessarily mimic that in embryonic hearts.
On the other hand, in the adult heart of mice overexpressing the Notch ligand Jagged-1 on cardiomyocytes, activated Notch limits adverse cardiac remodeling [11]. Notch actually impedes cardiomyocyte apoptosis as well as cardiac hypertrophy in adult mouse hearts subjected to pressure overload and fibrosis mediated by transforming growth factor-β and its effector connective tissue growth factor [11]. Hence, the Notch pathway acts on cardiac mesenchymal stromal cells (cardiac fibroblasts and precursor cells). It reduces the proliferation of myofibroblasts. It also stimulates the expansion of SCA1+ and NKx2-5+ cardiac precursor cells [11].
Overexpression of cyclin-A2 that enables the cell cycle progression through the G2–M checkpoint (Vol. 2, Chap. 2. Cell Growth and Proliferation) can engender cardiomyocyte proliferation [7]. Cyclin-D1 causes a sustained DNA synthesis, but abnormal patterns of multinucleation. Overexpressed cyclin-D2 supports regenerative proliferation.
In transgenic mice, overexpression of CDK2 kinase also transiently raises cardiomyocyte proliferation [7]. Less-differentiated and mononucleated cardiomyocytes abound.
The transcription factor and regulator of cardiogenesis Meis homeodomain-containing protein Meis1 acts as a modulator of cell proliferation after birth.
The genetic Meis1 deletion extends cardiomyocyte proliferation after birth and reactivates cell division in adult hearts [7].
The postnatal cardiomyocyte proliferation is controlled by many signaling cascades, such as the Hippo pathway, and modulated by microRNAs.
The Hippo kinase cassette intervenes in myogenesis, organ size control, and regeneration of the heart [12]. Kinases and scaffold proteins of the Hippo axis suppress cell proliferation. The Hippo pathway regulates the activity of TEA domain-containing transcription factors (TEAD1–TEAD4) mainly via phosphorylation of the transcriptional coactivators YAP1 and WWTR1 that activate TEAD factors (Vol. 4, Chap. 10. Other Major Types of Signaling Mediators).
The cardiomyocyte proliferation involves the activation of IGF1 receptor, subsequent PKB activation, and inactivation of GSK3β, which stabilizes β-catenin, a promoter cardiac development [7].
The main effectors of the Hippo pathway are the transcriptional coactivators YAP1 and YAP2 that derepress the genes activated by the Wnt–β Ctnn pathway. Genetic deletion of the upstream inactivating effectors (STK4, WW45 [Sav1], and LaTS1/2) causes cardiac hyperplasia [7]. On the other hand, transgenic mice overexpressing STK4, LaTS1, or LaTS2 die postnatally with dilated cardiomyopathy without compensatory ventricular myocyte hypertrophy.
1.2.8 Growth Factors and Cardiac Progenitor Proliferation in the Adult Heart
Various types of adult progenitor cells have been injected into damaged hearts or into coronary circulation, such as mesenchymal stromal cells, hematopoietic stem cells, endothelial progenitor cells, skeletal myoblasts, and cardiac progenitor cells to ensure cardiac protection, metabolism, contractility, proper remodeling, and neovascularization via stem cell paracrine signaling using growth factors, cytokines, and chemokines (FGF1–FGF2, HGF, IGF1, PDGF, VEGF, Nrg1, angiopoietin-1, IL6, and CXCL12), as well as differentiation and repopulation.
1.2.9 Heat Shock Proteins and Cardiac Repair
The member HSPa12b of the HSP70 family is predominately expressed in endotheliocytes of the heart, brain, lung, kidney, and adipose tissue, among others, but not in liver sinusoids [13]. In the heart and brain, the vascular endothelium of vessels of all sizes produces HSPa12b, whereas in lungs and adipose tissue, it is mainly synthesized in capillaries. It is required for angiogenesis, specifically in adhesion, migration, and tube formation.
The heat shock protein HSPa12b possesses 22 client proteins such as aryl hydrocarbon receptor nuclear translocator (ARNT). Among them, A-kinase-anchoring protein AKAP12 and podocalyxin-like protein PodxL are implicated in cell adhesion.
HSPa12b protects endotheliocytes against lipopolysaccharide-induced dysfunction and inflammation, as well as cerebral ischemia–reperfusion injury via the PI3K–PKB pathway possibly stimulated by angiopoietin-1 [13].
Transgenic mice expressing human HSPa12b specifically in endotheliocytes (without change in expression of other HSPs [i.e., HSPb5, HSP25, HSP60, HSP72, and HSP90]) have an improved cardiac function and remodeling assessed by left ventricular enlargement, wall thinning, and fibrosis, with a lower rate of cardiomyocyte apoptosis and higher capillary and arteriolar densities, from 1 to 4 weeks after myocardial infarction compared with wild-type mice [14]. The HSPa12b chaperone yields a protective and reparative response to ischemic insult in the heart.
Under ischemia, levels of proteins involved in cardiomyocyte survival and angiogenesis, such as NOS3 (without significant change of NOS2), Ang1, VEGF, and BCL2, are further elevated [14]. Angiopoietin-1 has an anti-inflammatory and protective actions on endotheliocytes via the PI3K–PKB axis, a decreased expression of cell adhesion molecules ICAM1 and VCAM1 and hence immunocyte recruitment, and an increased expression of antiapoptotic proteins such as BCL2. The VEGF factor augments NOS3 expression, thereby enhancing nitric oxide synthesis. The latter messenger acts against oxidative stress, inflammation, hypertrophy, and fibrosis, thereby reducing adverse cardiac remodeling. Protected cardiomyocytes, in turn, secrete VEGF and paracrine factors that protect endotheliocytes and support angiogenesis.
1.3 Molecular Chaperones in Cardiac Diseases
The continuous activity of cardiomyocytes requires the maintenance of proteostasis, that is, a stringent control of synthesis, folding, and turnover of proteins (Vol. 1, Chap. 5. Protein Synthesis), especially in sarcomeres, endoplasmic reticulum (ER), and mitochondria.
Molecular chaperones are devoted to regulating protein folding and maintaining the correct balance between synthesis and degradation. Molecular chaperones, heat shock proteins (HSP), prevent accumulation of damaged proteins either by supporting their refolding or by targeting them to proteasomal or autophagosomal degradation. In addition, chaperones participate in intracellular signaling, as they control conformational changes required for activation and deactivation of signaling mediators and their assembly in signalosomes.
In normal conditions, cells react to chemical and adverse mechanical stresses using molecular chaperones of the salvage machinery. However, high and persistent mechanical overload, oxidative stress, shifts in pH and temperature, as well as genetic mutations, can provoke accumulation of misfolded proteins. Insoluble aggregates of misfolded proteins that then form are toxic for the cell. Accumulation and aggregation of misfolded protein can be involved in cardiac diseases, such as dilated cardiomyopathy, arrhythmias, and heart failure [15].
Stimulation of chaperone synthesis has a cardioprotective role in ischemia–reperfusion injury, heart failure, and arrhythmias [15]. On the other hand, mutations in genes encoding chaperones generate different forms of cardiomyopathies.
The chaperone machinery consists of numerous proteins that cooperate. Chaperones constitute a large set of proteins that can be subdivided into three groups (Tables 1.3, 1.4, and 1.5) [15]:
1.
The general chaperones HSP90 and HSP70;
2.
The small chaperones (sHSP); and
3.
The ER chaperones
Table 1.3
Molecular chaperones in pathophysiology. (Part 1) General chaperones and cochaperones. (Source: [15])
Chaperone | Targets | Pathophysiological role |
|---|---|---|
HSP90 | Protein folding | Linkage to immature K 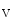 11.1 11.1 |
PKB–NOS3 axis | ↓ ischemia–reperfusion injury | |
Raf–ERK axis | ||
HSP70 | Protein folding | Production induced by ischemia |
Ubiquitination of misfolded proteins | Linkage to immature K 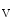 11.1 11.1 | |
Intracellular signaling | ||
HSP40 | Mitochondrial genesis | Avoids dilated cardiomyopathy |
CHIP (STUB1) | Unfolded proteins moved to proteasome | Limit infraction extension |
Table 1.4
Molecular chaperones in pathophysiology. (Part 2) Small chaperones. (Source: [15])
Chaperone | Targets | Pathophysiological role |
|---|---|---|
HSPb1 (HSP25) | Protein folding, aggregation | Protection against atrial fibrillation |
HSPb2 (HSP27) | Protein folding, aggregation | Reduction of ischemia–reperfusion damage |
HSPb5 | Desmin and titin folding | Reduction of oxidative stress Attenuation of apoptosis and necrosis Opposition to cardiac hypertrophy |
HSPb6 (HSP20) | PKB/BAD/Casp3 MAP3K5–JNK/P38 | Cardioprotection DCM (gene mutation) |
HSPb7 | Cytoskeletal protein folding (?) | Protection against atrial fibrillation DCM (gene SNP) |
HSPb8 | Autophagy | Reduction of infarct size |
HSP22 | BMPR2–MAP3K7–PI3K–PKB STAT3 | Opposition to fibrosis |
Melusin | FAK; ERK1/2, PKB–GSK3β | Preservation of contractility |
Table 1.5
Molecular chaperones in pathophysiology. (Part 3) Endoplasmic reticulum chaperones and stress response proteins. (Source: [15])
Chaperone | Targets | Pathophysiological role |
|---|---|---|
HSPa5 | Protein folding | Resistance to ischemia |
PDI | Protein folding | Reduction of infarct size |
DDIT3 (CHOP) | Apoptosis | Heart failure Cardiac hypertrophy, fibrosis |
XBP1 | ER chaperone synthesis | Dominant-negative XBP1 augments apoptosis |
ATF6 | ER chaperone synthesis | Production of HSPa5 and HSP90β1 Left ventricle protection upon pressure overload |
1.3.1 General Chaperones
The function of HSP90 and HSP70 is tightly controlled by cochaperones that regulate the specificity and dynamics of a chaperone reaction, as they recognize specific target proteins and control ATP binding and hydrolysis. Cochaperones also participate in the decision toward degradation of misfolded proteins.
For example, HSP70 binds to its substrate assisted by its cochaperone HSP40. The additional cochaperone HSP70/HSP90-organizing protein homolog (HOP)5 promotes the transfer of the substrate to HSP90 that enables conformational changes of the substrate. Other additional cochaperones, such as the HSP90 cochaperone P23, which is the cytosolic prostaglandin-E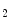 synthase PGeS3, and prolyl isomerase, regulate HSP90 ATPase activity and protein folding [15].
synthase PGeS3, and prolyl isomerase, regulate HSP90 ATPase activity and protein folding [15].
synthase PGeS3, and prolyl isomerase, regulate HSP90 ATPase activity and protein folding [15].1.3.2 Small Chaperones
Small chaperones (sHsp or HSPb) constitute a family of 11 members that can homo- and hetero-oligomerize. This polymerization can be regulated by transient phosphorylation in response to stress [15].
Small chaperones are devoid of ATPase activity. Nonetheless, they can prevent misfolded protein aggregation and regulate intracellular signaling, sometimes, in cooperation with HSP70 and HSP90.
1.3.3 Chaperones of the Endoplasmic Reticulum
The ER of cardiomyocytes consists of a vesicular compartment mainly in the perinuclear region involved in protein synthesis, and another compartment organized around myofibrils and T-tubules, which controls excitation–contraction coupling via calcium cycling. The two networks are interconnected.
Hypoxia, blood pressure overload, and drug-induced insults activate the ER chaperone machinery that maintains the proper balance between protein folding and degradation in the ER.
The ER chaperone machinery that assists protein folding and catalyzes disulphide bond formation includes:
Calnexin and calreticulin that control Ca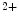 homeostasis;
homeostasis;
Chaperones HSPa5 and HSP90β1;
Protein disulphide isomerase; and
Thiol oxidoreductase-like protein ERP57.
These molecular chaperones act in concert with three ER membrane complexes, PERK, IRE1α, and ATF6, which activate the ER stress response [15]. In unstressed conditions, PERK (or eIF2α K3 kinase), IRN1 (or inositol-requiring Ser/Thr protein kinase and endoribonuclease IRe1 or IRe1α), and ATF6 bind to HSPa5 in the ER lumen, which inhibits their function. Upon ER stress, HSPa5 connects to misfolded proteins in the ER lumen, dissociating from PERK, IRN1, and ATF6 that become activated. The PERK (pancreatic eIF2α ER kinase) represses protein synthesis, as it phosphorylates the eukaryotic initiation factor-2α. Endoplasmic reticulum-to-nucleus signaling protein ERN1 generates a spliced XBP1 mRNA encoding a potent transcription factor that activates the expression of ER stress response genes. X-box-binding protein XBP1 upregulates genes involved in ubiquitin-dependent protein proteasomal degradation. Activating transcription factor ATF6 translocates to the Golgi body upon activation and undergoes cleavage by specific peptidases, yielding a soluble cytosolic transcription factor that enters the nucleus and induces further transcription of ER stress response genes.
Prolonged or extreme ER stress induces the expression of the transcription factor CHOP that triggers synthesis of proapoptotic proteins and ER-mediated cell death.
1.3.4 Protein Degradation and Autophagy
C-terminus of HSP70-interacting protein (CHIP) is a ubiquitin ligase highly expressed in the heart that, in cooperation with HSP70 and BCL2-associated athanogene (BAG) family molecular chaperone regulator BAG1, controls degradation of sarcomeric proteins by the proteasome [15].
In the absence of CHIP, ischemic damage leads to an increased infarct size. When misfolded proteins accumulate, proteic aggregates form that can activate autophagy by lysosomal hydrolases. Autophagy is observed in heart failure and ischemic heart disease [15].
The cochaperones BAG3 and HSPb8 operate in autophagic clearance in the heart (chaperone-assisted selective autophagy) [15]. Mutations of the Bag3 gene cause dilated cardiomyopathy.
1.3.5 Molecular Chaperones and Signal Transduction
Chaperones and scaffold proteins promote the formation of proteic complexes and stabilize the interaction of signaling components. The HSP90 substrates include the kinases Src, Raf, PKB, PI3K, and PDK1, as well as NOS3 [15]. Interaction of HSP90 with kinases involves the cochaperone protein Cdc37 (also called HSP90 chaperone protein kinase-targeting subunit).
Most signaling mediators are degraded upon inhibition of HSP90. The latter operates in VEGF-dependent NOS3 activation in endotheliocytes that leads to the disruption of the caveolin–NOS3 complex and promotes the NOS3–HSP90 association. The NOS3–HSP90 dimer recruits PKB that phosphorylates NOS3 (Ser1177) [15].
In addition to HSP90, sHSPs and cochaperones (e.g., HSPb6, HSPb8, melusin, and BAG3) regulate signaling pathways that control cardiomyocyte survival and calcium cycling. The small HSPb6 chaperone promotes cardiomyocyte survival via PKB and inhibition of the MAP3K5–JNK/P38MAPK modules. XS in response to β-adrenergic stimuli, HspB6 that binds to PP1 phosphatase, hence increasing phospholamban phosphorylation, is phosphorylated by PKA (Ser16), thereby activating SERCA2a pump [15].
Morgana and Melusin cooperate with HSP90 to regulate signal transduction in pathophysiological processes. Morgana is ubiquitously expressed, whereas Melusin is a striated muscle-specific chaperone [16]. Morgana controls genomic stability by regulating the centrosome cycle via the RoCK2 kinase. Melusin, or integrin-β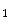 -binding protein Itgβ1BP2 organizes ERK signaling in cardiomyocytes and regulates cardiac hypertrophy in response to pressure overload.
-binding protein Itgβ1BP2 organizes ERK signaling in cardiomyocytes and regulates cardiac hypertrophy in response to pressure overload.
-binding protein Itgβ1BP2 organizes ERK signaling in cardiomyocytes and regulates cardiac hypertrophy in response to pressure overload.Melusin, in association with HSP90, promotes the organization of the Melusin signalosome that regulates ERK1, ERK2, and PKB signalings in response to different stresses. The Melusin signalosome comprises the scaffold and RhoGAPIQGAP1 that binds to FAK as well as the kinases of MAPK modules cRaf, MAP2K1, MAP2K2, ERK1, and ERK2.
1.3.6 Molecular Chaperones and Gene Mutations
Chaperones can tether to polypeptide variants encoded by mutated genes and support their refolding, hence restoring their function. Moreover, HSP90 can prevent transposon-induced genetic variations via piRNAs involved in transposon silencing [15].
The Hspb5 gene is mutated (R120G) in certain types of cardiomyopathies. The R157H mutation that affects the interaction of HSPb5 with titin can be detected in dilated cardiomyopathy. Conversely, overexpression of HSPb5 prevents formation of proteic aggregates of desmin variants that cause cardiomyopathies. A heterozygous Hspb6 point mutation (P20L) can also be identified in some cases of dilated cardiomyopathy.
1.3.7 Cardioprotective Chaperones
The mitochondrial chaperone HSP40 contributes to the prevention of dilated cardiomyopathy. Overexpression of HSPb1 ensures protection against pacing-induced cardiomyocyte damage, as it preserves sarcomeric constituents from proteolysis by calpain [15]. HSPb6–HSPb8 also protect against tachycardia-induced atriomyocyte remodeling.
1.4 Control of Cardiomyocyte Fate and Function
1.4.1 Nitric Oxide Synthases—Mechanochemotransduction
In cardiomyocytes, stretch and hence contraction trigger the production of reactive oxygen and nitrogen species that target Ca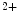 -signaling proteins ( mechanochemotransduction). This process involves nitric oxide synthases (NOS1 more than NOS3), NADPH oxidase NOx2, and Ca
-signaling proteins ( mechanochemotransduction). This process involves nitric oxide synthases (NOS1 more than NOS3), NADPH oxidase NOx2, and Ca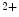 -calmodulin-dependent kinase CamK2 [17].
-calmodulin-dependent kinase CamK2 [17].
-signaling proteins ( mechanochemotransduction). This process involves nitric oxide synthases (NOS1 more than NOS3), NADPH oxidase NOx2, and Ca-calmodulin-dependent kinase CamK2 [17].The NOS1 subtype is situated closer to the Ca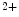 release sites, that is, to ryanodine receptors. The NOS1 isoform, but not NOS3, operates in afterload-induced Ca
release sites, that is, to ryanodine receptors. The NOS1 isoform, but not NOS3, operates in afterload-induced Ca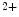 sparks via elevated ryanodine receptor sensitivity, thereby enhancing contractility to counter mechanical load [17].
sparks via elevated ryanodine receptor sensitivity, thereby enhancing contractility to counter mechanical load [17].
release sites, that is, to ryanodine receptors. The NOS1 isoform, but not NOS3, operates in afterload-induced Ca sparks via elevated ryanodine receptor sensitivity, thereby enhancing contractility to counter mechanical load [17].On the other hand, spontaneous Ca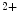 sparks during diastole can be arrhythmogenic. Both CamK2 and NOx2 also contribute to afterload-induced Ca
sparks during diastole can be arrhythmogenic. Both CamK2 and NOx2 also contribute to afterload-induced Ca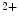 sparks. However, mechanotransduction launched by NOS1 and CamK2 does not depend on NOx2, as inhibition of NOS1 and CamK2, but not NOx2, in cardiomyocytes eliminates Ca
sparks. However, mechanotransduction launched by NOS1 and CamK2 does not depend on NOx2, as inhibition of NOS1 and CamK2, but not NOx2, in cardiomyocytes eliminates Ca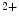 sparks, at least in a mouse model of familial hypertrophic cardiomyopathy with enhanced mechanotransduction [17].
sparks, at least in a mouse model of familial hypertrophic cardiomyopathy with enhanced mechanotransduction [17].
sparks during diastole can be arrhythmogenic. Both CamK2 and NOx2 also contribute to afterload-induced Ca sparks. However, mechanotransduction launched by NOS1 and CamK2 does not depend on NOx2, as inhibition of NOS1 and CamK2, but not NOx2, in cardiomyocytes eliminates Ca sparks, at least in a mouse model of familial hypertrophic cardiomyopathy with enhanced mechanotransduction [17].1.4.2 StIM1–Orai1-Mediated Store-Operated Calcium Entry
Cycle of Ca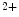 influx and outflux from the cytosol relies on Ca
influx and outflux from the cytosol relies on Ca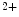 store refilling. The latter process is carried out by Ca
store refilling. The latter process is carried out by Ca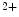 reuptake from the cytosol and activation of Ca
reuptake from the cytosol and activation of Ca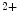 influx using the slow store-operated Ca
influx using the slow store-operated Ca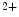 entry (SOCE) mechanism. The latter is delayed by more than 30 s after store depletion.
entry (SOCE) mechanism. The latter is delayed by more than 30 s after store depletion.
influx and outflux from the cytosol relies on Ca store refilling. The latter process is carried out by Ca reuptake from the cytosol and activation of Ca influx using the slow store-operated Ca entry (SOCE) mechanism. The latter is delayed by more than 30 s after store depletion.In cardiomyocytes, SOCE coexists with Ca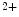 -induced Ca
-induced Ca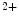 release (CICR) from the ER mediated by the couple formed by plasmalemmal voltage-gated Ca
release (CICR) from the ER mediated by the couple formed by plasmalemmal voltage-gated Ca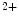 channels and ryanodine receptors on the apposed ER membrane.
channels and ryanodine receptors on the apposed ER membrane.
-induced Ca release (CICR) from the ER mediated by the couple formed by plasmalemmal voltage-gated Ca channels and ryanodine receptors on the apposed ER membrane.In response to binding of angiotensin-2 and endothelin-1 to their respective GPCRs, the newly produced inositol trisphosphate (IP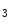 ) binds to its receptor (IP
) binds to its receptor (IP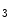 R) on the ER membrane, causing Ca
R) on the ER membrane, causing Ca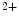 release from its main store.
release from its main store.
) binds to its receptor (IPR) on the ER membrane, causing Ca release from its main store.1.4.2.1 Involved Molecules
In cardiomyocyte, store-operated Ca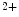 entry mediated by stromal interaction molecule StIM1 in conjunction with plasmalemmal Ca
entry mediated by stromal interaction molecule StIM1 in conjunction with plasmalemmal Ca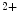 release-activated Ca
release-activated Ca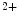 (CRAC) channels Orai1 and canonical transient receptor potential protein (TRPC) contributes to Ca
(CRAC) channels Orai1 and canonical transient receptor potential protein (TRPC) contributes to Ca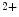 handling and hence excitation–contraction coupling.
handling and hence excitation–contraction coupling.
entry mediated by stromal interaction molecule StIM1 in conjunction with plasmalemmal Ca release-activated Ca (CRAC) channels Orai1 and canonical transient receptor potential protein (TRPC) contributes to Ca handling and hence excitation–contraction coupling.The StIM1–Orai1 complex mediates highly Ca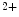 -selective and nonvoltage-gated current. On the other, the StIM1–TRPC complex conveys Ca
-selective and nonvoltage-gated current. On the other, the StIM1–TRPC complex conveys Ca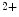 -selective and -nonselective store-operated currents, as TRPC is a relatively nonselective cation channel conducting inward currents carried by both monovalent and divalent cations (e.g., Na
-selective and -nonselective store-operated currents, as TRPC is a relatively nonselective cation channel conducting inward currents carried by both monovalent and divalent cations (e.g., Na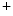 and Ca
and Ca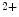 ) [18]. Among TRPCs, TRPC1, TRPC3, TRPC4, and TRPC6 are implicated in SOCE-mediated hypertrophic signaling in cardiomyocytes.
) [18]. Among TRPCs, TRPC1, TRPC3, TRPC4, and TRPC6 are implicated in SOCE-mediated hypertrophic signaling in cardiomyocytes.
-selective and nonvoltage-gated current. On the other, the StIM1–TRPC complex conveys Ca-selective and -nonselective store-operated currents, as TRPC is a relatively nonselective cation channel conducting inward currents carried by both monovalent and divalent cations (e.g., Na and Ca) [18]. Among TRPCs, TRPC1, TRPC3, TRPC4, and TRPC6 are implicated in SOCE-mediated hypertrophic signaling in cardiomyocytes.The Orai1–StIM1 complex localizes in particular to the transverse tubule of skeletal myocytes [19]. Its rate of activation is regulated by the RyR1 receptor.
Stromal Interaction Molecule
The STIM family comprises two proteins (StIM1–StIM2). The most widely studied StIM1, a ubiquitous type-1 membrane protein, predominantly localizes to the ER membrane where it acts as an Ca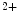 sensor that detects changes in ER Ca
sensor that detects changes in ER Ca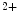 concentration. In response to Ca
concentration. In response to Ca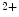 depletion in the ER, Ca
depletion in the ER, Ca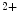 dissociates from StIM1 that then oligomerizes and translocates to subplasmalemmal punctae.
dissociates from StIM1 that then oligomerizes and translocates to subplasmalemmal punctae.
sensor that detects changes in ER Ca concentration. In response to Ca depletion in the ER, Ca dissociates from StIM1 that then oligomerizes and translocates to subplasmalemmal punctae.StIM1
At rest, StIM1 dimerizes. In the absence of store depletion, StIM1 clusters in cytosolic regions rather than at portions near the plasma membrane. Upon ER Ca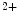 depletion, StIM1 oligomerizes and moves to regions close to the plasma membrane (ER–PM junctions) and links to Orai1 (mainly) or TRPC channels. Oligomerization of StIM1 depends on phosphorylation.
depletion, StIM1 oligomerizes and moves to regions close to the plasma membrane (ER–PM junctions) and links to Orai1 (mainly) or TRPC channels. Oligomerization of StIM1 depends on phosphorylation.
depletion, StIM1 oligomerizes and moves to regions close to the plasma membrane (ER–PM junctions) and links to Orai1 (mainly) or TRPC channels. Oligomerization of StIM1 depends on phosphorylation.StIM1 is also a microtubule plus-end-binding protein serving in microtubule extension. However, in contrast to all other cytosolic plus-end-tracking proteins, StIM1 is attached to the ER membrane. It binds to microtubule-associated protein of the RP/EB family MAPRE1 at sites where polymerizing microtubule ends contact the ER [20]. ER tubule elongates with the MAPRE1+ end of a growing microtubule. Therefore, microtubule growth-dependent concentration of StIM1 in the ER membrane enables ER remodeling.
Stanniocalcin-1 is a secreted glycoproteic auto- or paracrine regulator of calcium and phosphate homeostasis. It prevents Ca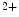 uptake. Stanniocalcin Stc2 is an ER stress protein as a component of the unfolded protein response, the expression of which is induced by oxidative stress and hypoxia. It colocalizes with StIM1 and behaves as a SOCE inhibitor [21].
uptake. Stanniocalcin Stc2 is an ER stress protein as a component of the unfolded protein response, the expression of which is induced by oxidative stress and hypoxia. It colocalizes with StIM1 and behaves as a SOCE inhibitor [21].
uptake. Stanniocalcin Stc2 is an ER stress protein as a component of the unfolded protein response, the expression of which is induced by oxidative stress and hypoxia. It colocalizes with StIM1 and behaves as a SOCE inhibitor [21].In addition, StIM1 oligomerization and SOCE are precluded by the ER-resident protein disulfide isomerase PDIa3 [22].
Other modulators of StIM1 and Orai1 include EF-hand calcium-binding domain-containing protein EFCaB4b, or CRAC regulator CRACR2a. The cytosolic Ca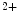 sensor CRACR2a stabilizes CRAC channels, as it promotes StIM–Orai binding at low Ca
sensor CRACR2a stabilizes CRAC channels, as it promotes StIM–Orai binding at low Ca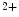 concentrations. The ternary Orai1–StIM1–CRACR2a complex dissociates at elevated Ca
concentrations. The ternary Orai1–StIM1–CRACR2a complex dissociates at elevated Ca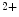 levels [23].
levels [23].
sensor CRACR2a stabilizes CRAC channels, as it promotes StIM–Orai binding at low Ca concentrations. The ternary Orai1–StIM1–CRACR2a complex dissociates at elevated Ca levels [23]. Another ER integral membrane protein Surf4 (Surfeit locus protein-4 or simply Surfeit-4) binds to StIM1 and modulates StIM1-dependent SOCE [24]. This modulator impedes StIM1 clustering upon store depletion.
The ubiquitous ER membrane protein, SOCE-associated regulatory factor SARAF [25, 26]: (1) connects to the StIM1–Orai1 activation region (SOAR) of StIM1 domain (which activates Orai1); (2) facilitates slow Ca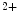 -dependent inactivation of Orai1 via the C-terminal inhibitory domain (CTID) of StIM1 (which hinders spontaneous StIM1 clustering and full Orai1 activation in the absence of store depletion and determines access of SARAF to the SOAR motif to regulate both FCDI and SCDI); and (3) protects cells from calcium overload.
-dependent inactivation of Orai1 via the C-terminal inhibitory domain (CTID) of StIM1 (which hinders spontaneous StIM1 clustering and full Orai1 activation in the absence of store depletion and determines access of SARAF to the SOAR motif to regulate both FCDI and SCDI); and (3) protects cells from calcium overload.
-dependent inactivation of Orai1 via the C-terminal inhibitory domain (CTID) of StIM1 (which hinders spontaneous StIM1 clustering and full Orai1 activation in the absence of store depletion and determines access of SARAF to the SOAR motif to regulate both FCDI and SCDI); and (3) protects cells from calcium overload.At rest, SARAF binds to the SOAR sequence of the StIM1 protein. Activation of Orai1 requires dissociation of SARAF from the SOAR domain that then can tether to Orai1.
StIM2
In mice, global deletion of the STIM1 gene is lethal within 2 days after birth, whereas lack of StIM2 enables survival until 4–5 weeks [18]. The StIM2 subtype has a different N-terminus with respect to StIM1. Once it is linked to Orai1, StIM2 causes both store-dependent and -independent Ca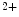 entry.
entry.
entry.StIM1L
In addition, the high-molecular weight splice variant StIM1L also lodges in cardiomyocytes [18]. It forms a permanent cluster with actin and colocalizes with Orai1 [27]. The unique behavior of the CRAC flux in skeletal myocytes may result from the coupling of Orai1 channels with StIM1L [19].
Orai
The ORAI family includes three members (Orai1–Orai3). The StIM1 protein also contributes to store-independent Ca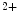 entry via the arachidonic acid-regulated Ca
entry via the arachidonic acid-regulated Ca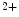 channel that is a heteromer consisting of both Orai1 and Orai3 channels, whereas the classical SOCE flux relies on StIM1 connected to an Orai1 homohexamer [18].
channel that is a heteromer consisting of both Orai1 and Orai3 channels, whereas the classical SOCE flux relies on StIM1 connected to an Orai1 homohexamer [18].
entry via the arachidonic acid-regulated Ca channel that is a heteromer consisting of both Orai1 and Orai3 channels, whereas the classical SOCE flux relies on StIM1 connected to an Orai1 homohexamer [18].At rest, Orai1 homodimerizes or homotetramerizes. Upon activation, it hexamerizes. Orai1 is a highly Ca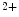 -selective, inward-rectifying channel prominently regulated (inactivated) by Ca2+ ion. The half time of the 2 modes of inactivation, fast (FCDI) and slow (SCDI) Ca2+-dependent inactivation, ranges from 10 to 100 ms and 2 to 3 ms, respectively [26].The former depends on StIM1 and calmodulin binding to the Orai1 channel [25].
-selective, inward-rectifying channel prominently regulated (inactivated) by Ca2+ ion. The half time of the 2 modes of inactivation, fast (FCDI) and slow (SCDI) Ca2+-dependent inactivation, ranges from 10 to 100 ms and 2 to 3 ms, respectively [26].The former depends on StIM1 and calmodulin binding to the Orai1 channel [25].
-selective, inward-rectifying channel prominently regulated (inactivated) by Ca2+ ion. The half time of the 2 modes of inactivation, fast (FCDI) and slow (SCDI) Ca2+-dependent inactivation, ranges from 10 to 100 ms and 2 to 3 ms, respectively [26].The former depends on StIM1 and calmodulin binding to the Orai1 channel [25].1.4.2.2 SOCE Goal, Regulation, and Function
In response to IP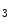 R- and/or RyR-mediated Ca
R- and/or RyR-mediated Ca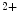 release from sarcoplasmic reticulum stores, SOCE enables Ca
release from sarcoplasmic reticulum stores, SOCE enables Ca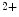 influx from the extracellular space and a sustained increase in cytosolic Ca
influx from the extracellular space and a sustained increase in cytosolic Ca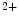 concentration to regulate gene transcription (Vol. 4, Chap. 11. Signaling Pathways). SOCE is also aimed at rapidly refilling the depleted store. Although StIM1 and Orai1 alone can reconstitute SOCE activity, other proteins are involved (e.g., SERCA).
concentration to regulate gene transcription (Vol. 4, Chap. 11. Signaling Pathways). SOCE is also aimed at rapidly refilling the depleted store. Although StIM1 and Orai1 alone can reconstitute SOCE activity, other proteins are involved (e.g., SERCA).
R- and/or RyR-mediated Ca release from sarcoplasmic reticulum stores, SOCE enables Ca influx from the extracellular space and a sustained increase in cytosolic Ca concentration to regulate gene transcription (Vol. 4, Chap. 11. Signaling Pathways). SOCE is also aimed at rapidly refilling the depleted store. Although StIM1 and Orai1 alone can reconstitute SOCE activity, other proteins are involved (e.g., SERCA).Once SOCE is activated, it is subjected to various regulatory processes that determine the duration and magnitude of the Ca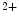 influx. In particular, the StIM1–Orai1 complex is regulated by the SGK1 and AMPK kinases that may reduce unnecessary SOCE when energy reserves are low [18]. On the other hand, in pulmonary arterial smooth myocytes, PDGF activates the PKB–TOR pathway and, subsequently, enhances the SOCE process and cell proliferation [28].
influx. In particular, the StIM1–Orai1 complex is regulated by the SGK1 and AMPK kinases that may reduce unnecessary SOCE when energy reserves are low [18]. On the other hand, in pulmonary arterial smooth myocytes, PDGF activates the PKB–TOR pathway and, subsequently, enhances the SOCE process and cell proliferation [28].
influx. In particular, the StIM1–Orai1 complex is regulated by the SGK1 and AMPK kinases that may reduce unnecessary SOCE when energy reserves are low [18]. On the other hand, in pulmonary arterial smooth myocytes, PDGF activates the PKB–TOR pathway and, subsequently, enhances the SOCE process and cell proliferation [28].The SOCE process is specifically involved in the IP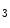 -mediated α-adrenergic signaling, but does not transmit the β-adrenergic message [18].
-mediated α-adrenergic signaling, but does not transmit the β-adrenergic message [18].
-mediated α-adrenergic signaling, but does not transmit the β-adrenergic message [18].1.4.2.3 Pathophysiological Role of SOCE
SOCE and Metabolic Perturbations
Hyperglycemia is associated with a decrease in total ER Ca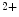 content and StIM1 level at least in endotheliocytes [18]. In smooth myocytes, SOCE is inhibited by hyperglycemia. In platelets from type-2 diabetic patients, SOCE is reduced because of attenuated association between StIM1 and Orai1, TRPC1, and TRPC6 channels.
content and StIM1 level at least in endotheliocytes [18]. In smooth myocytes, SOCE is inhibited by hyperglycemia. In platelets from type-2 diabetic patients, SOCE is reduced because of attenuated association between StIM1 and Orai1, TRPC1, and TRPC6 channels.
content and StIM1 level at least in endotheliocytes [18]. In smooth myocytes, SOCE is inhibited by hyperglycemia. In platelets from type-2 diabetic patients, SOCE is reduced because of attenuated association between StIM1 and Orai1, TRPC1, and TRPC6 channels.The SOCE process may be regulated by  Glc
Glc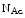 attachment (O-GlcNAcylation). A sustained posttranslational protein modification by
attachment (O-GlcNAcylation). A sustained posttranslational protein modification by  linked
linked 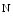 acetylglucosamine contributes to the adverse effects of hyperglycemia [18]. Glucosamine, an amine sugar that selectively fuels the hexosamine synthesis pathway, mimickes the effects of hyperglycemia [18]. Inhibition of glucose entry into the hexosamine synthesis pathway suppresses the effects of hyperglycemia on SOCE. Angiotensin-2-induced increase in cytosolic Ca
acetylglucosamine contributes to the adverse effects of hyperglycemia [18]. Glucosamine, an amine sugar that selectively fuels the hexosamine synthesis pathway, mimickes the effects of hyperglycemia [18]. Inhibition of glucose entry into the hexosamine synthesis pathway suppresses the effects of hyperglycemia on SOCE. Angiotensin-2-induced increase in cytosolic Ca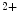 level is attenuated in cardiomyocytes by increasing the
level is attenuated in cardiomyocytes by increasing the 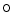 Glc
Glc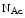 level [18]. O-GlcNAcylation targets sites that are frequently subjected to phosphorylation. O-GlcNAcylation of StIM1 actually prevents its phosphorylation and lowers StIM1-associated subplasmalemmal puncta formation and subsequently SOCE-mediated Ca
level [18]. O-GlcNAcylation targets sites that are frequently subjected to phosphorylation. O-GlcNAcylation of StIM1 actually prevents its phosphorylation and lowers StIM1-associated subplasmalemmal puncta formation and subsequently SOCE-mediated Ca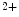 flux [18].
flux [18].
Glc attachment (O-GlcNAcylation). A sustained posttranslational protein modification by linked acetylglucosamine contributes to the adverse effects of hyperglycemia [18]. Glucosamine, an amine sugar that selectively fuels the hexosamine synthesis pathway, mimickes the effects of hyperglycemia [18]. Inhibition of glucose entry into the hexosamine synthesis pathway suppresses the effects of hyperglycemia on SOCE. Angiotensin-2-induced increase in cytosolic Ca level is attenuated in cardiomyocytes by increasing the Glc level [18]. O-GlcNAcylation targets sites that are frequently subjected to phosphorylation. O-GlcNAcylation of StIM1 actually prevents its phosphorylation and lowers StIM1-associated subplasmalemmal puncta formation and subsequently SOCE-mediated Ca flux [18].SOCE and Arrhythmias
On the other hand, Orai1 and Orai3 may be linked to arrhythmias in both atrial and ventricular myocytes [18]. The TRPC channel is produced in pacemaker cells of the sinoatrial node. Moreover, elevated Orai-mediated SOCE can initiate atrial and ventricular arrhythmias.
SOCE and Cardiac Hypertrophy
StIM1 is necessary and sufficient in the occurrence of cardiomyocyte hypertrophy due to the pressure overload in the adult heart. Calcium-mediated cardiomyocyte hypertrophy such as that primed by angiotensin-2 depends on SOCE, but neither on Ca 1.2a nor Na
1.2a nor Na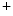 –Ca
–Ca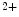 exchanger [18]. Both TRPC3 and TRPC6 contribute to the development of angiotensin-2-induced hypertrophy. A TRPC4-dependent sustained increase in cytosolic Ca
exchanger [18]. Both TRPC3 and TRPC6 contribute to the development of angiotensin-2-induced hypertrophy. A TRPC4-dependent sustained increase in cytosolic Ca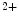 activates the PP3–NFAT pathway implicated in cardiac hypertrophy. Endothelin-1 activates SOCE, upregulates TRPC1 expression, and provokes NFAT nuclear translocation and cardiomyocyte hypertrophy. In addition, the concentration of the large StIM1 splice variant StIM1L rises in response to hypertrophic stimuli.
activates the PP3–NFAT pathway implicated in cardiac hypertrophy. Endothelin-1 activates SOCE, upregulates TRPC1 expression, and provokes NFAT nuclear translocation and cardiomyocyte hypertrophy. In addition, the concentration of the large StIM1 splice variant StIM1L rises in response to hypertrophic stimuli.
1.2a nor Na–Ca exchanger [18]. Both TRPC3 and TRPC6 contribute to the development of angiotensin-2-induced hypertrophy. A TRPC4-dependent sustained increase in cytosolic Ca activates the PP3–NFAT pathway implicated in cardiac hypertrophy. Endothelin-1 activates SOCE, upregulates TRPC1 expression, and provokes NFAT nuclear translocation and cardiomyocyte hypertrophy. In addition, the concentration of the large StIM1 splice variant StIM1L rises in response to hypertrophic stimuli.SOCE and Ischemia–Reperfusion Injury
Cardiomyocyte
During ischemia–reperfusion injury, calcium overload was assumed to result from the coupling of Na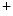 –H
–H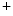 exchanger with Na
exchanger with Na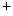 –Ca
–Ca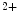 exchanger operating in its reversal mode and, to a lesser extent, Ca
exchanger operating in its reversal mode and, to a lesser extent, Ca 1.2a channel [18]. In fact, SOCE inhibition protects the heart against Ca
1.2a channel [18]. In fact, SOCE inhibition protects the heart against Ca overload induced by acute ischemia–reperfusion injury.
overload induced by acute ischemia–reperfusion injury.
–H exchanger with Na–Ca exchanger operating in its reversal mode and, to a lesser extent, Ca1.2a channel [18]. In fact, SOCE inhibition protects the heart against Ca overload induced by acute ischemia–reperfusion injury.Hypoxia stimulates StIM1 accumulation at subplasmalemmal punctae possibly due to the action of ROS. However, the accompanying acidosis uncouples the StIM1–Orai1 complex, thereby preventing Ca overload [18].
overload [18].
overload [18].Upon reperfusion, the acidosis is quickly resolved and SOCE may be again activated. In addition, the Ca concentration in the sarcoplasmic reticulum decays rapidly during reperfusion.
concentration in the sarcoplasmic reticulum decays rapidly during reperfusion.
concentration in the sarcoplasmic reticulum decays rapidly during reperfusion.In cardiomyocytes subjected to ischemia–reperfusion injury, overexpressed TRPC3 increases apoptosis and calpain activation with respect to control cardiomyocytes [18].
Endotheliocyte
Pharmacological inhibition of SOCE abolishes Ca overload in cardiac microvascular endotheliocytes subjected to ischemia–reperfusion injury [18].
overload in cardiac microvascular endotheliocytes subjected to ischemia–reperfusion injury [18].
overload in cardiac microvascular endotheliocytes subjected to ischemia–reperfusion injury [18].1.4.3 Sphingosine 1-Phosphate
Circulating sphingosine 1-phosphate signals via RhoA GTPase that promotes cardiomyocyte survival and PLCε to prime the phosphorylation (activation) of protein kinase-D1, thereby ensuring cardioprotection against ischemia–reperfusion injury [29]. The PKD1 kinase phosphorylates (inhibits) the cofilin phosphatase slingshot homolog SSH1, thereby impeding cofilin-2 translocation to mitochondria in response to oxidative stress or ischemia–reperfusion injury. Cofilin-2 links to the proapoptotic protein BAX. Therefore, S1P preserves mitochondrial integrity upon oxidative stress and supports cardiomyocyte survival.
1.4.4 Forkhead Box Transcription Factors
The member of the forkhead box family of transcription factors FoxO1 is involved in oxidative stress response, immunity, pluripotency in embryonic stem cells, and regulation of cell metabolism, proliferation, and death (Vol. 4, Chap. 10. Other Major Types of Signaling Mediators).
The FoxO1 factor promotes gluconeogenesis, as it stimulates synthesis of hepatic glucose 6-phosphatase and phosphoenolpyruvate carboxykinase [30]. In skeletal muscle, FoxO1 raises expression of pyruvate dehydrogenase kinase, thereby decreasing glucose oxidation. In the heart, FoxO1 regulates glucose and fatty acid metabolism, as it targets the genes that encode pyruvate dehydrogenase kinase PDK4 and nitiric oxide synthase NOS2 [30].
In the heart, cellular insulin-stimulated glucose uptake through the GluT1 (basal glucose uptake) and mainly GluT4 (stimulated glucose uptake) and glycolysis leads to the formation of pyruvate that undergoes oxidation in the mitochondria to yield ATP. In cardiomyocytes, insulin resistance is linked to PDK4 overexpression, hence with lowered glucose and heightened fatty acid oxidation due to nuclear FoxO1 action [30].
The FoxO1 factor also contributes to the regulation of the supply and oxidation of fatty acids. Dietary fatty acids are delivered to the heart from lipoprotein lipase-mediated lipolysis of triglyceride-rich lipoproteins as well as by albumin-bound fatty acids. The uptake of fatty acids by cardiomyocytes across the plasma membrane is mediated by three protein-fatty acid translocases: plasmalemmal and mitochondrial glutamate oxaloacetate transaminase GOT2, fatty acid transport protein (FATP), and the ScaRb3 receptor. AcylCoA synthase enhances fatty acid uptake. The FoxO1 agent is implicated in lipid supply and storage, as its nuclear localization is correlated with an elevated plasmalemmal density of ScaRb3.
The FoxO1 factor is involved in the regulation of the oxidative stress response. It increases expression of antioxidant genes such as those that encode superoxide dismutase and the stress sensor GADD45, thereby promoting ROS scavenging and preventing DNA damage [30]. It collaborates with SIRT1, hence activating expression of antioxidant genes such as those that encode 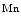 SOD and catalase.
SOD and catalase.
SOD and catalase.In embryonic stem cells, FoxO1 can maintain their pluripotency, a site acts on promoters of the OCT4 and SOX2 genes.
1.4.5 Regulation by 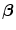 -Adrenoceptors
-Adrenoceptors
β-adrenoceptor regulation of cardiac contraction and gene expression is mediated by protein kinase-A. The PKA activity is also controlled by several other messengers acting via Gs-coupled receptors. The velocity and specificity of β-AR signaling are determined by the localization of the PKA holoenzyme and associated A-kinase anchoring proteins as well as the spatiotemporal pattern of cAMP signaling.
The second messenger cAMP is small diffusible molecule. The cAMP–PKA couple is organized into signaling complexes organized by A-kinase anchoring proteins that tether inactive PKA holoenzymes and other signaling molecules and distributed in discrete compartments in cardiomyocytes [31]. From these cellular foci, specific cAMP signals propagate to determined subcellular loci. In cardiomyocytes, AKAP6 anchors PKA to multiple subcellular compartments, including the nucleus and nuclear membrane [32]. In the nucleus, AKAP6 also connects to PDE4d3 that controls the cAMP level.
On the sarcoplasmic reticulum in cardiomyocytes, PKA regulates the calcium cycling for excitation–contraction coupling that is dysfunctional in heart diseases. The direct activation of adenylate cyclases or application of a cAMP analog provokes a much slower and smaller increase in PKA activity than stimulated adrenergic receptors [31]. The signaling primed by adrenoceptors has a preferential access to the sarcoplasmic reticulum, the major calcium store. Calcium ions are released through ryanodine receptors and taken up through sarco(endo)plasmic reticulum Ca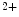 ATPase. The triggered cAMP–PKA signaling leads to phosphorylation of these ion carriers and/or associated regulatory proteins, thereby raising Ca
ATPase. The triggered cAMP–PKA signaling leads to phosphorylation of these ion carriers and/or associated regulatory proteins, thereby raising Ca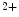 flux through these channels to adapt cardiac contraction.
flux through these channels to adapt cardiac contraction.
ATPase. The triggered cAMP–PKA signaling leads to phosphorylation of these ion carriers and/or associated regulatory proteins, thereby raising Ca flux through these channels to adapt cardiac contraction.In the absence of cAMP, the PKA holoenzyme is a heterotetramer consisting of two catalytic subunits bound and inhibited by a dimer of regulatory subunits. Activation of adenylate cyclases increases intracellular cAMP concentration. This second messenger dissociates (activates) the catalytic subunits.
Then, PKA phosphorylates many substrates in various subcellular compartments, such as the ion channels involved in excitation–contraction coupling, sarcolemmal Ca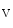 1.2a and sarcoplasmic reticulum RyR2, the SERCA inhibitor phospholamban (Pln), and the sarcomeric components cardiac myosin-binding protein-C (cMyBPc) and troponin-I (cTnnI; Sect. 4.1.1).
1.2a and sarcoplasmic reticulum RyR2, the SERCA inhibitor phospholamban (Pln), and the sarcomeric components cardiac myosin-binding protein-C (cMyBPc) and troponin-I (cTnnI; Sect. 4.1.1).
1.2a and sarcoplasmic reticulum RyR2, the SERCA inhibitor phospholamban (Pln), and the sarcomeric components cardiac myosin-binding protein-C (cMyBPc) and troponin-I (cTnnI; Sect. 4.1.1).In the nucleus of cardiomyocytes, PKA targets the transcription factors of the CREB family and class-2 histone deacetylases HDAC4 and HDAC5.
The cAMP concentration depends on its production by adenylate cyclases and degradation by phosphodiesterases (PDE1–PDE4 and PDE8) in cardiomyocytes. Members of the cAMP-specific PDE4 family (PDE4a–PDE4b and PDE4d encoded by three genes) are major inhibitors of the cAMP messenger. At the nuclear envelope, PDE4d complexes with AKAP6. It is involved in the regulation of cardiomyocyte hypertrophy. The loss of PDE4d can cause dilated cardiomyopathy, at least in mice. AKAP-anchored PDE4 controls the extent of PKA released upon βAR stimulation and transferred into the nucleus from a cytosolic pool [32].
In the cytoplasm, phosphatases PP1 and PP2 contribute to the termination of PKA action, whereas only PP1 acts in the nucleus [32].
In adult rat ventriculomyocytes, β-adrenoceptor stimulation raises quickly the cytoplasmic and nuclear cAMP concentration, whereas PKA activity elevation is prompt in the cytoplasm, but slower in the nucleus [32]. Similar slow kinetics of nuclear PKA activation is observed upon adenylate cyclase activation or phosphodiesterase inhibition. Pulse stimulation causes maximal PKA activation and phosphorylation of myosin-binding protein-C in the cytoplasm, but has a slight impact on PKA activation and phosphorylation of cAMP response element-binding protein in the nucleus.
The nuclear PKA activity is temporally dissociated from that of nuclear cAMP elevation and cytoplasmic PKA activation. Once PKA holoenzyme is activated in the cytoplasm, its catalytic subunit translocates to the nucleus [32].
In cardiomyocytes, the upstream inhibitors PDE3 and PDE4 and downstream inhibitors PP1, PP2, and PP3 can participate in the cytoplasmic and nuclear PKA response to βAR stimulation.
The PDE3 and PDE4 enzymes represent the major PDE activity in cardiomyocyte nuclei. They primarily localize at the nuclear envelope. The PDE4d subtype controls the nuclear as well as cytosolic PKA activity, whereas PDE3 has no effect [32].
The PP1 and PP2 phosphatases contribute differently to PKA target sites in the cytoplasm and nucleus. They counteract the action of the βAR–cAMP–PKA axis, especially the phosphorylation of mediators of the electromechanical coupling, but PP3 has no effect [32]. The PP3 phosphatase may play a minor role when PP1 and PP2 are active. However, PP3 intervenes in pathogenesis, especially cardiac adverse hypertrophy resulting from pressure overload. In particular, PP3 dampens β AR effect on Pln in spontaneously hypertensive rats. Moreover, it is detected in the nucleus of cardiomyocytes in the diseased myocardium, but not in the case of healthy hearts [32].
In addition, cAMP can also activate 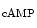 GEFs (RapGEF3–RapGEF4) at the plasma membrane and nuclear and perinuclear compartment area in cardiomyocytes [32]. These effectors increase nuclear Ca
GEFs (RapGEF3–RapGEF4) at the plasma membrane and nuclear and perinuclear compartment area in cardiomyocytes [32]. These effectors increase nuclear Ca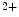 concentration as well as CamK2 dependent nuclear export of HDAC5 and subsequent derepression of the MEF2 transcription factor. Unlike CamK2- and PKD-mediated phosphorylation, PKA phosphorylates HDAC5 in the nucleus, thereby causing its nuclear retention and the repression of the hypertrophic gene program.
concentration as well as CamK2 dependent nuclear export of HDAC5 and subsequent derepression of the MEF2 transcription factor. Unlike CamK2- and PKD-mediated phosphorylation, PKA phosphorylates HDAC5 in the nucleus, thereby causing its nuclear retention and the repression of the hypertrophic gene program.
GEFs (RapGEF3–RapGEF4) at the plasma membrane and nuclear and perinuclear compartment area in cardiomyocytes [32]. These effectors increase nuclear Ca concentration as well as CamK2 dependent nuclear export of HDAC5 and subsequent derepression of the MEF2 transcription factor. Unlike CamK2- and PKD-mediated phosphorylation, PKA phosphorylates HDAC5 in the nucleus, thereby causing its nuclear retention and the repression of the hypertrophic gene program.A sustained βAR activation, as that occurring during intense and prolonged physical exercise or after myocardial injury, increases nuclear PKA activity and CREB phosphorylation [32]. Transcription factors of the CREB family can contribute to βAR-dependent maladaptive remodeling.
1.5 Interactions Between Cardiac Cell Populations
The heart is composed of different cell types that interact to maintain the cardiac and bodily homeostasis, in addition to the dynamical partnership created by the nervous, renal, and cardiovascular system.
The largest cardiac volume is occupied by cardiomyocytes. Cardiofibroblasts have the highest density. Other cardiac cell types include nodal cells, epicardiocytes, endocardial and vascular endotheliocytes, coronary smooth myocytes, leukocytes, myofibroblasts, adipocytes, mesenchymal stem cells, and sympathetic and parasympathetic neurons (Vol. 5, Chap. 6. Heart Wall).
Cardiac cell types need a well-handled communication to optimize signaling resulting from numerous messengers. The intercellular communication encompasses direct between-cell contact, cell–matrix interaction, and long-range signals, using chemical (short peptides, proteins, lipids, nucleotides, and RNAs) and electrochemical (ions) cues. In addition to secreted molecules, cells release vesicles that contain proteins, RNAs, and sometimes DNA.
Interactions between different cardiac cell types enable proper myocardial contractility, sufficient perfusion, adequate myocardial stiffness, and controlled functioning of immunity. However, heterotypic intercellular interactions are also involved in disease progression.
Interactome refers to gene and protein interactions within a pathway as well as between distinct axes (genic and proteic interactomes), as well as cell communications inside a population of a given cell type (isotypic interactions, i.e., connections between cells of the same bulk phenotype) and between different populations of cell types (heterotypic signaling, i.e., data transmission between cells of different phenotypes) in a spatiotemporal regulated manner and in a given physiological or pathological context (cellular interactome; Tables 1.6 and 1.7).
Table 1.6
Examples of messengers involved in interactions between cardiac cells
Source | Target | Messengers |
|---|---|---|
CMC | CFB | ANP, BNP, TGFβ, CTGF, IL1β/6, miR133a |
EC | Nrg1, CSF3, TNFSF1 | |
CFB | CMC | CNP, ET1, CTGF, FGF2, PDGF, TGFβ, |
TNFSF1, IL6/10/17/33, LIF, CT1 | ||
EC | CMC | ET1 |
SMC | MiR126/143/145 | |
SMC | CFB | CTGF |
Leukocytes | CMC | TNFSF1, IL17 |
CFB | TNFSF1 | |
Stem cells | CMC | ATP |
EC | SHh |
Table 1.7
Short- and long-distance bidirectional communication between different cell types in the heart. Cardiac myocytes (85 % of the cardiac mass) and fibroblasts (∼ 2/3 the total number of cardiac cells), vascular endothelial and smooth muscle cells, and epicardiocytes constitute the main cell populations in the healthy adult heart. Extra- and intracellular signals that regulate cardiomyocyte proliferation during the postnatal life include growth factors and microRNAs. In diseases, these heterocellular connections reorganize and initiate adaptive and maladaptive responses. Exosomes secreted from stem and progenitor cells yield beneficial effects in the injured myocardium. (Source: [33])
Type of communication | Mediators | Role |
|---|---|---|
Healthy heart | ||
Myocyte–endotheliocyte | Neuregulin ROS | Angiogenesis Cardiomyocyte survival, cardiomyocyte hypertrophy |
Fibroblast–immunocyte | Cytokines | Feedback control of fibroblast and leukocyte activation |
Diseased heart | ||
Myocyte–endotheliocyte | Neuregulin ROS | Angiogenesis Cardiomyocyte survival, cardiomyocyte hypertrophy |
Myocyte–macrophage (M1) | Cytokines DAMPs | |
Myocyte–fibroblast | MicroRNAs | Tissue repair |
lncRNAs | Tissue stiffness Contractile dysfunction | |
Fibroblast–stem cell | Cytokines | Suppression of myofibroblast differentiation |
Fibroblast–adipocyte | Adipokines Activin-A | Myofibroblast differentiation Fibrosis |
Fibroblast–macrophage | Cytokines | |
Endotheliocyte–macrophage (M2) | Neovascularization Myocardial infarction healing | |
A subpopulation of a given cell type such as fibroblast with its given functional characteristics can influence the activity of other isotypic subpopulations, in addition to other cell types.
Acute ischemic injury and maladaptive cardiomyocyte hypertrophy are the most common acute and chronic insults to the heart. After cardiac injury, the spatial arrangement of cells is disrupted and the cellular composition is altered, as different cell populations are temporally recruited to the heart. These cell types interfere using many signaling pathways.
1.5.1 Examples of Signaling Messengers
After cardiac injury, multiple signaling pathways, such as the Notch, Wnt, TGFβ, and BMP axes, are activated.
The Notch-primed signaling exerts a negative feedback on the Wnt–β Ctnn pathway; the intracellular domain of Notch receptor can bind to Wnt signaling effectors such as Disheveled, thereby inhibiting Wnt signaling.
Calcium ions are major second messengers in both the cardiac pump and vasculature. Localized changes in Ca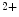 ions encompass waves, oscillations, sparks, sparklets, puffs, transients, and flashes, which can correspond to different triggering events.
ions encompass waves, oscillations, sparks, sparklets, puffs, transients, and flashes, which can correspond to different triggering events.
ions encompass waves, oscillations, sparks, sparklets, puffs, transients, and flashes, which can correspond to different triggering events.1.5.1.1 Hedgehog
During myocardial ischemia, the Hedgehog pathway promotes neovascularization, thereby increasing cardiomyocyte survival. The canonical Hedgehog pathway activates gene transcription via transcription factors of the Gli family upon derepression of the GPCR Smoothened.
In neonatal rat ventriculomyocytes, sonic Hedgehog activates Gli1 via Smoothened and Gi proteins [34]. The Gli1 transcription factor then targets a subset of 37 cardiomyocyte-specific genes, such as those encoding mediators of the PKA and nucleotide pathways.
1.5.1.2 Wnt
The Wnt–βCtnn axis with the transmembrane -frizzled receptors and related antagonists is used during cardiogenesis and cardiac injury response. The Wnt messengers constitute a family of 19 lipophilic proteins that bind to ten types of transmembrane receptors of the frizzled family and 2 types of coreceptors lipoprotein receptor-related proteins LRP5 and LRP6 (Vol. 3, Chap. 10. Morphogen Receptors). They trigger β-catenin-dependent canonical and noncanonical pathways.
The Wnt signaling is hindered by two categories of antagonists. Secreted frizzled-related protein (sFRP1–sFRP5) either directly binds to extracellular Wnt or competitively to Fz receptor, thereby preventing Wnt binding to Fz receptor in both cases. Dickoppf proteins (Dkk1–Dkk3) competitively preclude Wnt binding to LRP5 and LRP6 coreceptors.
The Wnt family members Wnt2, Wnt5a, Wnt5b, Wnt7b, Wnt9a, and Wnt11 as well as their antagonists sFRP1, sFRP2, and Dkk3 are expressed in the normal heart [35]. Mesenchymal cells in cardiac valves and subsets of endothelial and smooth muscle cells scattered throughout the myocardium respond to Wnt (Table 1.8). The Wnt morphogens regulate interactions between fibroblasts and other cardiac myocyte and nonmyocyte cell types.
Table 1.8
Expression of Wnt messengers and antagonists in the normal heart and after ischemia (Source: [35]). The secreted frizzled (Fz)-related protein (SFRP) family comprises five glycoproteins in humans (sFRP1–sFRP5) that act as extracellular signaling ligands that can bind Wnt proteins and Fz receptors. The interaction between sFRP and Wnt proteins prevents the latter from binding Fz receptors. Signaling by Wnt mesengers can be downregulated by the formation of an inhibitory complex with the frizzled receptors. The Dickkopf (Wnt signaling inhibitor) family of secreted protein encompasses four members (Dkk1–Dkk4) that antagonize Wnt–βCtnn signaling, as they interact with the Wnt coreceptors LRP5 and LRP6. They are also high-affinity ligands for the transmembrane proteic modulators of Wnt signaling Kremen-1 and -2
Normal heart | Ischemia |
|---|---|
Involved molecules | |
Wnt2/5a–5b/7b/9a/11 | Wnt1/2/4/7/10b/11 |
sFRP1/2 | sFRP1/2/4 |
Dkk3 | Dkk1/Dkk2 |
Cells responsive to canonical Wnt signaling | |
Valve mesenchymal cells | Epicardial-derived cells of subepicardial space |
Subsets of smooth myocytes | Fibroblasts, smooth myocytes and endotheliocytes in the injury region |
Endotheliocytes | |
Endotheliocytes possess most types of frizzled receptors (Fz1–Fz2, Fz4–Fz7, and Fz9–Fz10). The Wnt1, Wnt3a, and Wnt5a proteins regulate endotheliocyte proliferation and migration. The sFRP molecules exert both pro- and antiangiogenic effects. Subtype sFRP1 promotes migration and tube formation, but reduces endotheliocyte proliferation [35]. In hearts of patients with dilated cardiomyopathy and coronary artery disease, sFRP3 and sFRP4 levels are elevated [35].
1.5.1.3 Signal Transducer and Activator of Transduction
Signal transducers and activators of transcription constitute a family of transcription factors (STAT1–STAT4, STAT5a–STAT5b, and STAT6) that are all expressed in the myocardium. They homo- and heterodimerize.
STAT Family
The STAT1 agent is activated in ischemia–reperfusion injury. It upregulates the expression of caspase-1, TNFSF6, and TNFRSF6a, thereby promoting apoptosis in cardiomyocytes [36].
The STAT2 factor heterodimerize with the STAT1 agent. The STAT3 factor targets genes encoding proteins and microRNAs. It intervenes especially in cardiac physiology and pathophysiology. The STAT4 factor modulates inflammation in autoimmune and infectious myocarditis [36].
Both STAT5 and STAT6 participate in effects of the renin–angiotensin–aldosterone axis. They are rapidly phosphorylated during angiotensin-2 stimulation in the nonischemic region and bind to the STAT consensus sequence in the angiotensinogen promoter, thereby causing a sustained autocrine activation of angiotensin-2-mediated signaling.
STAT3
The STAT3 factor targets genes encoding proteins and microRNAs. It is involved in mitochondrial energy production and in cell survival, proliferation and differentiation, and metabolism in cardiomyocytes, fibroblasts, endotheliocytes, progenitor cells, and various types of inflammatory cells, as well as oxidative stress and protective and stress-induced adverse cardiac remodeling [36].
The STAT3 factor participates in intercellular communication between cardiomyocytes, endotheliocytes, fibroblasts, and cardiac progenitor cells, as well as infiltrated immunocytes (Table 1.9).
Table 1.9
Intercellular communication relying on the STAT3 factor. The STAT3 factor is activated by numerous liganded receptors (Source: [36])
Cell type | Targeted cellular function |
|---|---|
Cardiomyocyte | Sarcomere organization (miR199a) |
Cardiomyocyte architecture (PRMT1–ADMA) | |
Secretome | |
Survival, growth | |
Oxidative stress (ROS) | |
Fibroblast | Secretome |
Extracellular matrix | |
Survival, proliferation, differentiation | |
Endotheliocyte | Adhesion |
Survival, proliferation, differentiation (apoptosis and reduced proliferation and migration in PPCM due to 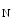 Prl and miR146a) Prl and miR146a) | |
Oxidative stress (ROS) | |
Progenitor cell | Survival, proliferation, differentiation |
Immunocyte | Secretome |
Differentiation | |
Mediators | |
Receptors | GP130 (IL6R), IL10R, CCR2, CSF3R, EpoR, Nrg1–HER, VEGFR, leptin R, AT  |
Cytokines | EPo, VEGF, Opn, Tsp1 |
MicroRNAs | MiR21/146a/199a |
ADMA asymmetric dimethylarginine, EPo erythropoietin, miR microRNA, Opn osteopontin, 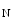 Prl antiangiogenic and proapoptotic N-terminal 16-kDa prolactin fragment, PPCM peripartum and postpartum cardiomyopathies, PRMT arginine
Prl antiangiogenic and proapoptotic N-terminal 16-kDa prolactin fragment, PPCM peripartum and postpartum cardiomyopathies, PRMT arginine 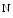 methyltransferase, ROS reactive oxygen species, Tsp1 thrombospondin-1, VEGF vascular endothelial growth factor
methyltransferase, ROS reactive oxygen species, Tsp1 thrombospondin-1, VEGF vascular endothelial growth factor
Prl antiangiogenic and proapoptotic N-terminal 16-kDa prolactin fragment, PPCM peripartum and postpartum cardiomyopathies, PRMT arginine methyltransferase, ROS reactive oxygen species, Tsp1 thrombospondin-1, VEGF vascular endothelial growth factorSTAT3 Signaling
The STAT3 factor is activated by multiple receptors. Its activity depends on dimerization and posttranslational modifications (acetylation [Lys49, Lys87, and Lys685] and phosphorylation [Ser727]). It is phosphorylated by PKCδ, PKCε, ERK1, ERK2, P38MAPK, CDK5, DAPK3, and TOR kinases, thereby potentiating the STAT3 transcriptional activity via recruitment of transcriptional cofactors, such as the P300 and CBP HATs.
The related signaling cascades are associated with positive and negative feedback loops. It participates in the production of the vasodilators, nitric oxide and prostacyclin. It mediates proangiogenic signaling via VEGF and erythropoietin.
Suppressor of cytokine signaling forms a negative feedback of STAT signaling. In particular, SOCS3 transcription is upregulated by STAT3 [36]. Both SOCS1 and SOCS3 interact with various JaK proteins or the GP130 receptor, thereby hindering STAT phosphorylation. Both PTPn6 and PTPn11 also target cytokine receptors and dephosphorylate JaK proteins, thereby impeding STAT3 phosphorylation.
Proteic inhibitors of activated STAT (PIAS1 and PIAS3) preclude DNA binding by dimerized phosphorylated STAT1 and STAT3, thereby diminishing transcriptional activation of STAT target genes. Protein Tyr phosphatase receptor PTPRt dephosphorylates STAT3, thereby, controlling target gene expression and cellular localization.
STAT3 can inhibit gene transcription. Acetylation (Lys685) involved in STAT3 interaction with DNA methyltransferase-1 causes methylation (silencing) of various gene promoters [36]. Deacetylation by histone deacetylases (e.g., HDAC1 and HDAC4) leads to STAT3 degradation.
MicroRNA-199a that alters the sarcomere organization is transcriptionally suppressed by STAT3 [36]. MicroRNA-199a targets the ubiquitin conjugases UbE2i and UbE2g1, thereby disturbing the protein turnover by the ubiquitin–proteasome axis and subsequently reducing expression of the genes that encode the sarcomeric α- and β-myosin heavy chain.
STAT3 in Cardiomyocyte–Endotheliocyte Communication
STAT3 is involved in the communication between cardiomyocytes and the myocardial vasculature. In cardiomyocytes, STAT3 provokes VEGFa expression in particular upon stimulation by IL6 and erythropoietin, especially in ischemia [36].
In endotheliocytes, STAT3 contributes to the expression of cytokines (e.g., IL6), chemokines (e.g., CCL2 and CX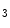 CL1), and cell adhesion molecules (ICAM1 and VCAM1). It promotes the differentiation of cardiac progenitor cells into endotheliocytes.
CL1), and cell adhesion molecules (ICAM1 and VCAM1). It promotes the differentiation of cardiac progenitor cells into endotheliocytes.
CL1), and cell adhesion molecules (ICAM1 and VCAM1). It promotes the differentiation of cardiac progenitor cells into endotheliocytes.Juxta- and paracrine communication between cardiomyocytes and between cardiomyocytes and endotheliocytes also relies on neuregulin-1 and colony-stimulating factor CSF3. The CSF3–STAT3 signaling improves cardiomyocyte survival and vascularization after myocardial infarction [36].
In addition, STAT3 antagonizes arginine  methyltransferase PRMT1 that increases the synthesis of asymmetric dimethylarginine (ADMA), an inhibitor of NO synthesis and promoter of ROS production in endotheliocytes [36]. The PRMT1 enzyme is activated in response to disturbed protein turnover and accumulation of damaged proteins, thereby severely damaging the cardiomyocyte structure and function and simultaneously impairing the endothelium in a paracrine manner, and hence ultimately priming initiation and progression of heart failure.
methyltransferase PRMT1 that increases the synthesis of asymmetric dimethylarginine (ADMA), an inhibitor of NO synthesis and promoter of ROS production in endotheliocytes [36]. The PRMT1 enzyme is activated in response to disturbed protein turnover and accumulation of damaged proteins, thereby severely damaging the cardiomyocyte structure and function and simultaneously impairing the endothelium in a paracrine manner, and hence ultimately priming initiation and progression of heart failure.
methyltransferase PRMT1 that increases the synthesis of asymmetric dimethylarginine (ADMA), an inhibitor of NO synthesis and promoter of ROS production in endotheliocytes [36]. The PRMT1 enzyme is activated in response to disturbed protein turnover and accumulation of damaged proteins, thereby severely damaging the cardiomyocyte structure and function and simultaneously impairing the endothelium in a paracrine manner, and hence ultimately priming initiation and progression of heart failure.The endothelial differentiation capacity of resident cardiac SCA1+ progenitor cells that express both the erythropoietin and chemokine CCR2 receptors requires STAT3 [36]. Cardiomyocyte-derived erythropoietin, production and secretion of which depends on STAT3, influences the expression of CCL2. In addition, the secretion of cytokines and chemokines from cardiac cells, such as IL11, CT1, and LIF, engenders endothelial differentiation of cardiac progenitor cells via the GP130–STAT3 axis. The mobilization of bone marrow endothelial progenitor cells into the blood circulation relies on an IL10–STAT3 pathway [36].
Interference between cardiomyocytes and bone marrow-derived mesenchymal stem cells affects the myogenic conversion [36]. The reduction of miR124 in bone marrow-derived mesenchymal stem cells generated by cardiomyocytes increases expression of STAT3 and STAT3-associated myocytic markers, such as atrial natriuretic peptide, troponin-T, and α-myosin heavy chain, as well as the cardiac potassium channel currents.
STAT3 in Cardiomyocyte–Fibroblast Communication
Fibroblasts and cardiomyocytes synthesize various matrix constituents as well as matrix-regulatory enzymes (matrix metallopeptidases and their inhibitors TIMPs). In fibroblasts, STAT3 promotes cell survival and proliferation, as well as the synthesis of matrix components (e.g., collagen). STAT3 is a major player in communication between cardiomyocytes and fibroblasts.
In cardiofibroblasts, STAT3 regulates not only the synthesis of genes encoding matrix constituents but also that of cytokines, as well as connective tissue growth factor, thrombospondin-1, tissue inhibitor of metallopeptidases TIMP1, osteopontin, tenascin-C, and plasminogen activator inhibitor-1 (or serpin-E1) [36].
Cardiac stress factors, such as ischemia, angiotensin-2, and pressure overload, induce the expression of IL6 family cytokines (i.e., IL6, IL11, leukemia inhibitory factor [LIF], oncostatin-M [OSm], and cardiotrophin-1 [CT1], among others) in both cardiomyocytes and fibroblasts. Communication between cardiomyocytes and fibroblasts relies on the IL6Rα–GP130 receptor. Other stress factors such as β-adrenoceptor agonists provoke production of IL6 cytokines only in cardiofibroblasts, but not in cardiomyocytes [36].
1.5.1.4 Tumor-Necrosis Factor TNFSF1
The proinflammatory cytokine tumor-necrosis factor-α (TNFSF1) connects to its cognate TNFR1 (TNFRSF1a) and TNFR2 (TNFRSF1b) receptors on cardiomyocytes.
In mice, acute stimulation with a low TNFSF1 dose (10 μg/kg) raises cytosolic calcium transient amplitude and cardiac contractility [37]. In mice, TNFSF1 overexpression causes dilated cardiomyopathy. On the other hand, exposure of adult cardiomyocytes to TNFSF1 during 12 h protects against hypoxic injury.
The Ras interaction/interference protein RIN1, afadin, and Ras association domain-containing protein family member RASSF1a modulate TNFSF1 signaling in cardiomyocytes. RASSF1a does not interact with TNFRSF1b. This adaptor facilitates the recruitment of TRADD that links TNFRSF1a and RIPK1, and TRAF2 to form the TNFRSF1a complex. It is required for signal transmission from the TNFRSF1a complex to effectors, such as cytoplasmic phospholipase-A2 and protein kinase-A, but not calcium–calmodulin-dependent kinase CamK2. It enables activation of Ca 1.2a and RyR2 channels [37]. In addition, RASSF1a also complexes with plasma membrane calcium ATPase PMCA4, a calcium extrusion pump located in caveolae, as is TNFRSF1a.
1.2a and RyR2 channels [37]. In addition, RASSF1a also complexes with plasma membrane calcium ATPase PMCA4, a calcium extrusion pump located in caveolae, as is TNFRSF1a.
1.2a and RyR2 channels [37]. In addition, RASSF1a also complexes with plasma membrane calcium ATPase PMCA4, a calcium extrusion pump located in caveolae, as is TNFRSF1a.However, TNFSF1 is a proapoptotic cytokine. The TNFSF1–TNFRSF1a couple alters mitochondrial function and Ca handling and provokes cell death. In acute ischemia, TNFSF1 activates caspase-8 that alters mitochondrial functioning, increasing production of ROS, and causes Ca
handling and provokes cell death. In acute ischemia, TNFSF1 activates caspase-8 that alters mitochondrial functioning, increasing production of ROS, and causes Ca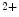 leak from the sarcoplasmic reticulum [38].
leak from the sarcoplasmic reticulum [38].
handling and provokes cell death. In acute ischemia, TNFSF1 activates caspase-8 that alters mitochondrial functioning, increasing production of ROS, and causes Ca leak from the sarcoplasmic reticulum [38].In murine adult ventriculomyocytes exposed to TNFSF1, caspase-8 is activated, thereby increasing the mitochondrial production of ROS and priming poly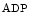 ribose polymerase PARP1 activation and hence production of adenosine diphosphoribose (
ribose polymerase PARP1 activation and hence production of adenosine diphosphoribose (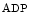 ribose) and subsequent poly(ADP-ribosyl)ation of proteins. In particular, oxidative stress stimulates Ca
ribose) and subsequent poly(ADP-ribosyl)ation of proteins. In particular, oxidative stress stimulates Ca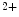 -permeable TRPM2 channel that can be activated by the intracellular second messenger
-permeable TRPM2 channel that can be activated by the intracellular second messenger 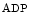 ribose, in addition to nicotinamide adenine dinucleotide (NAD
ribose, in addition to nicotinamide adenine dinucleotide (NAD ) and cyclic adenosine diphosphate–ribose (cADPR) [38]. The TRPM2 channel conveys a nonspecific cationic current that mediates TNFSF1-induced ventriculomyocyte death. Its inhibition thus protects against apoptosis.
) and cyclic adenosine diphosphate–ribose (cADPR) [38]. The TRPM2 channel conveys a nonspecific cationic current that mediates TNFSF1-induced ventriculomyocyte death. Its inhibition thus protects against apoptosis.
< div class='tao-gold-member'>
ribose polymerase PARP1 activation and hence production of adenosine diphosphoribose (ribose) and subsequent poly(ADP-ribosyl)ation of proteins. In particular, oxidative stress stimulates Ca-permeable TRPM2 channel that can be activated by the intracellular second messenger ribose, in addition to nicotinamide adenine dinucleotide (NAD) and cyclic adenosine diphosphate–ribose (cADPR) [38]. The TRPM2 channel conveys a nonspecific cationic current that mediates TNFSF1-induced ventriculomyocyte death. Its inhibition thus protects against apoptosis.Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


