Mutations in the bone morphogenetic protein receptor type 2 ( BMPR2 ) gene and the activin receptor–like kinase 1 ( ALK1 ) gene have been reported in heritable pulmonary arterial hypertension (HPAH) and idiopathic pulmonary arterial hypertension (IPAH). However, the relation between clinical characteristics and each gene mutation in IPAH and HPAH is still unclear, especially in childhood. The aim of this study was to determine, in a retrospective study, the influence and clinical outcomes of gene mutations in childhood IPAH and HPAH. Fifty-four patients with IPAH or HPAH whose onset of disease was at <16 years of age were included. Functional characteristics, hemodynamic parameters, and clinical outcomes were compared in BMPR2 and ALK1 mutation carriers and noncarriers. Overall 5-year survival for all patients was 76%. Eighteen BMPR2 mutation carriers and 7 ALK1 mutation carriers were detected in the 54 patients with childhood IPAH or HPAH. Five-year survival was lower in BMPR2 mutation carriers than mutation noncarriers (55% vs 90%, hazard ratio 12.54, p = 0.0003). ALK1 mutation carriers also had a tendency to have worse outcome than mutation noncarriers (5-year survival rate 64%, hazard ratio 5.14, p = 0.1205). In conclusion, patients with childhood IPAH or HPAH with BMPR2 mutation have the poorest clinical outcomes. ALK1 mutation carriers tended to have worse outcomes than mutation noncarriers. It is important to consider aggressive treatment for BMPR2 or ALK1 mutation carriers.
Pulmonary arterial hypertension (PAH) is a progressive, devastating disease. In the absence of treatment, PAH leads to death, with median survival of 2.8 years for adults. However, the median survival of children with PAH without treatment is <10 months. Furthermore, although <25% of adults with idiopathic PAH (IPAH) are acutely responsive to vasodilator testing, >40% of children with IPAH are responders. Thus, the prognosis and clinical features of IPAH in children may be different from those in adults. The clinical features of IPAH and heritable PAH (HPAH) in children, however, remain unclear. Bone morphogenetic protein receptor type 2 ( BMPR2 ) has been identified as a primary gene for HPAH. Heterozygous mutation of the activin receptor–like kinase 1 gene ( ALK1 ) has also been demonstrated in patients with hereditary hemorrhagic telangiectasia (HHT) in association with PAH. In addition, our group reported 5 ALK1 mutations in PAH children. Even rarer mutations in endoglin have been identified in patients with PAH, predominantly with coexistent HHT. In contrast, we reported the first nonsense mutation of SMAD8 and 2 missense mutations of BMPR1B in patients with IPAH. The differences in clinical outcomes in each gene mutation carrier in IPAH and HPAH are still unclear. There are several studies referring to this relation, but no report has investigated it in childhood IPAH and HPAH. In this study, we attempted to screen for other genes associated with these genes in pediatric patients and conducted a follow-up survey to clarify the clinical features and interrelations between gene mutations and outcomes in pediatric IPAH and HPAH.
Methods
Fifty-seven unrelated patients aged ≤15 years at diagnosis with PAH were selected from 19 hospitals throughout Japan and China ( Figure 1 ). There was no duplication in this selection process. Some of these patients have been described in previous reports. Written informed consent was obtained from guardians of all study subjects in accordance with the Declaration of Helsinki. We assessed each patient by clinical history, physical examination, current therapy, 6-minute walking distance, and a review of medical records through March 2011.
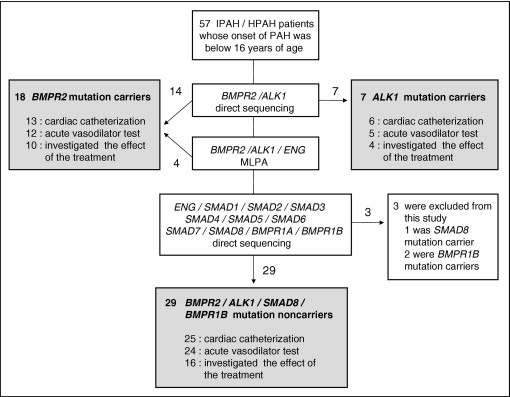
Diagnosis of IPAH and HPAH was made through clinical evaluation, chest x-ray, electrocardiography, echocardiography, and cardiac catheterization on the basis of current international consensus criteria (mean pulmonary artery pressure >25 mm Hg at rest or >30 mm Hg during exercise). Patients with PAH associated with other diseases such as portal hypertension, congenital heart disease, or persistent pulmonary hypertension of the newborn were systematically excluded from this study by trained cardiologists using a diagnostic algorithm. A significant acute response to nitric oxide and/or epoprostenol was defined by the following 3 criteria: (1) a >20% decrease in mean pulmonary artery pressure, (2) no change or an increase in cardiac index, and (3) no change or a decrease in the ratio of pulmonary vascular resistance to systemic vascular resistance.
The BMPR2 and ALK1 coding regions and exon-intron boundaries were amplified from patients’ genomic deoxyribonucleic acid. Amplified products were purified using the QIAquick polymerase chain reaction purification method (Qiagen, Hilden, Germany) and screened using bidirectional direct sequencing with an ABI 3130xl DNA Analyzer (Applied Biosystems, Foster City, California). Fourteen BMPR2 mutations and 7 ALK1 mutations were identified by direct sequencing ( Figure 1 ). Regarding BMPR2 and ALK1 , mutation-negative samples, multiplex ligation–dependent probe amplification (MLPA) was used to detect exonic deletions or duplications in BMPR2 , ALK1 , and endoglin. MLPA was performed according to the manufacturer’s instructions using a SALSA MLPA HHT/PPH1 probe set (MRC-Holland, Amsterdam, The Netherlands). MLPA analysis revealed that 4 of 36 patients had exonic deletions in BMPR2 ( Figure 1 ). In patients with no mutations in BMPR2 or ALK1 , all coding exons and adjacent intronic regions for endoglin, SMAD1 , SMAD2 , SMAD3 , SMAD4 , SMAD5 , SMAD6 , SMAD7 , SMAD8 , BMPR1A , and BMPR1B were amplified using a polymerase chain reaction. Polymerase chain reaction–amplified products were purified and directly sequenced like BMPR2 and ALK1 . One SMAD8 nonsense mutation and 2 BMPR1B missense mutations were detected, as described previously. They were excluded from this analysis because the number of cases representing each mutation was too small. No mutations were identified in endoglin, SMAD1 , SMAD2 , SMAD3 , SMAD4 , SMAD5 , SMAD6 , SMAD7 , or BMPR1A in 29 patients who had no mutations in BMPR2 , ALK1 , SMAD8 , or BMPR1B ( Figure 1 ). When a mutation was detected, we confirmed that it was not present in >200 healthy controls by direct sequencing.
Clinical features are expressed as mean ± SD, median (interquartile range), or number of patients (percentage), as appropriate. Univariate comparisons between ALK1 mutation carriers, BMPR2 mutation carriers, and mutation noncarriers were done using 1-way analysis of variance or the median test for continuous measures and the chi-square test for categorical measures. Death rate is presented as the number per person-year, and the difference between gene mutations was evaluated using a Poisson regression model.
A Kaplan-Meier overall survival curve was constructed to demonstrate the overall survival difference among mutation groups and compared using the log-rank test. Cox proportional-hazards regression modeling was performed to examine the relation between mutations and all-cause death. Similar survival analyses were performed by gender.
A p values <0.05 was considered significant. Statistical analyses were performed using JMP for Windows version 9 (SAS Institute Inc., Cary, North Carolina).
Results
From January 1, 1995, to March 31, 2011, 54 pediatric patients with IPAH or HPAH, corresponding to 24 male and 30 female patients, were included in this analysis. Eighteen (33%) were BMPR2 mutation carriers, 7 (13%) were ALK1 mutation carriers, and the rest were without these mutations ( Figure 1 , Table 1 ). The average age at diagnosis was 8.5 years, and there was no significant difference in ages among the 3 groups. Familial cases in ALK1 mutation carriers were largest in these groups. No significant differences were seen in brain natriuretic peptide or 6-minute walking distance. Cardiac catheterization was performed in 44 of 54 patients ( Figure 1 , Table 2 ). Forty-one of 54 patients were subjected to acute vasodilator response testing, 15 of whom were responders. There were no significant differences among the 3 groups in terms of hemodynamic characteristics or response to vasoreactivity testing ( Table 2 ).
| Variable | ALK1 Mutation Carriers (n = 7) | BMPR2 Mutation Carriers (n = 18) | Mutation Noncarriers (n = 29) | All Patients (n = 54) | p Value |
|---|---|---|---|---|---|
| Male gender | 3 (43%) | 10 (56%) | 11 (38%) | 24 (44%) | 0.496 |
| Family history of PAH | 4 (57%) | 3 (17%) | 3 (10%) | 10 (19%) | 0.012 |
| Age at onset (years) | 9.3 ± 4.2 | 9.1 ± 3.7 | 8.0 ± 4.0 | 8.5 ± 3.9 | 0.589 |
| Brain natriuretic peptide (pg/ml) ⁎ | 94.8 (7.8–752.0) | 159.2 (18.8–1,451.5) | 39.6 (12.2–117.1) | 57.0 (12.8–335.9) | 0.366 |
| 6-minute walking distance (m) ⁎ | 525 ± 176 | 379 ± 93 | 448 ± 113 | 442 ± 126 | 0.125 |
⁎ Although the period of 6-minute walking distance and brain natriuretic peptide measurement depended on the patient, we selected the newest data through March 2011. Brain natriuretic peptide could be measured in 12 BMPR2 mutation carriers, 6 ALK1 mutation carriers, and 22 mutation noncarriers and 6-minute walking distance in 8 BMPR2 mutation carriers, 5 ALK1 mutation carriers, and 15 mutation noncarriers.
| Variable | ALK1 Mutation Carriers (n = 6) | BMPR2 Mutation Carriers (n = 13) | Mutation Noncarriers (n = 25) | All Patients (n = 44) | p Value |
|---|---|---|---|---|---|
| Mean pulmonary artery pressure (mm Hg) | 61.8 ± 18.2 | 69.8 ± 19.7 | 62.1 ± 21.8 | 64.3 ± 20.6 | 0.529 |
| Right atrial pressure (mm Hg) | 6.7 ± 4.5 | 7.5 ± 4.5 | 6.6 ± 2.9 | 6.8 ± 3.6 | 0.758 |
| Cardiac index (L/min/m 2 ) | 3.7 ± 0.8 | 2.8 ± 1.1 | 3.3 ± 0.9 | 3.2 ± 1.0 | 0.196 |
| Total pulmonary resistance (Wood units/m 2 ) | 14.8 ± 4.3 | 26.3 ± 14.2 | 21.6 ± 11.9 | 21.9 ± 12.0 | 0.321 |
| Pulmonary vascular resistance (Wood units/m 2 ) | 17.1 ± 10.4 | 23.9 ± 14.6 | 17.6 ± 10.5 | 19.1 ± 11.6 | 0.411 |
| Pulmonary artery wedge pressure (mm Hg) | 9.0 ± 2.8 | 9.0 ± 2.9 | 9.0 ± 2.6 | 9.0 ± 2.7 | 0.999 |
| Acute vasodilator responder | 1/5 (20%) | 4/12 (33%) | 10/24 (42%) | 15/41 (37%) | 0.646 |
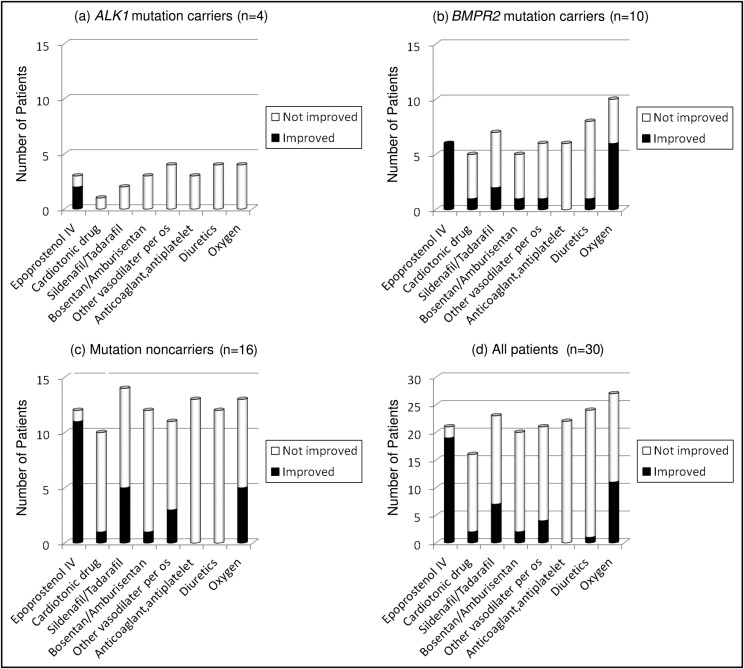
There were no significant differences in current therapy among the 3 groups ( Table 3 ). As shown in Figure 2 , intravenous epoprostenol strongly improved functional class in all groups. Sildenafil, vasodilators except sildenafil or tadalafil and bosentan or ambrisentan, and oxygen also improved functional class moderately. The difference of each drug effect among each group was not significant (data not shown).
| Variable | ALK1 Mutation Carriers (n = 4) | BMPR2 Mutation Carriers (n = 10) | Mutation Noncarriers (n = 23) | All Patients (n = 37) | p Value |
|---|---|---|---|---|---|
| Intravenous epoprostenol | 2 (50%) | 6 (60%) | 13 (57%) | 21 (57%) | 0.781 |
| Cardiotonic drugs | 1 (25%) | 4 (40%) | 9 (39%) | 14 (38%) | 0.709 |
| Sildenafil/tadalafil | 3 (75%) | 8 (80%) | 17 (74%) | 28 (76%) | 0.485 |
| Bosentan/ambrisentan | 4 (100%) | 7 (70%) | 15 (65%) | 26 (70%) | 0.273 |
| Other vasodilators per os | 4 (100%) | 8 (80%) | 14 (61%) | 26 (70%) | 0.179 |
| Anticoagulant or antiplatelet agents | 3 (75%) | 6 (60%) | 16 (70%) | 25 (68%) | 0.654 |
| Diuretics | 2 (50%) | 6 (60%) | 16 (70%) | 24 (65%) | 0.134 |
| Oxygen | 3 (75%) | 10 (100%) | 21 (91%) | 34 (92%) | 0.310 |
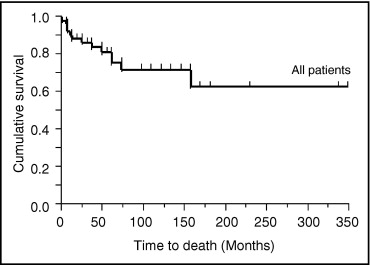
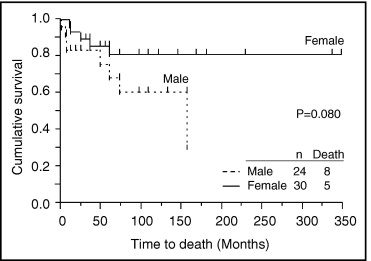
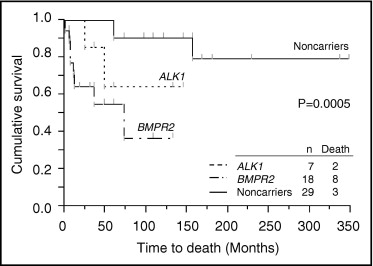
Eight of 18 BMPR2 mutation carriers died, and the causes of death were all directly related to PAH. Two of 7 ALK1 mutation carriers died of PAH. Three of 29 mutation noncarrriers died, and the causes of death were related to PAH. The rate of death was worst in BMPR2 mutation carriers ( Table 4 ). Overall 5- and 10-year survival of all patients was 76% and 72%, respectively ( Figure 3 ). As shown in Figure 4 , male patients’ outcomes tended to be worse than female patients’ outcomes. In Figure 5 , overall mortality was worst in BMPR2 mutation carriers. Five- and 10-year survival was lower in BMPR2 mutation carriers than mutation noncarriers (55% and 37% vs 90% and 90%, respectively). ALK1 mutation carriers also had a tendency to have worse outcomes than mutation noncarriers (5- and 10-year survival rates were 64%). A multivariate Cox proportional-hazards model for time to death indicated that BMPR2 mutation carriers had significantly worse outcome than mutation noncarriers ( Table 5 ). The difference in outcomes between ALK1 mutation carriers and mutation noncarriers was marginally significant. No significant differences were observed in time to death among the 3 subgroups of BMPR2 mutation carriers ( Figure 6 ).
| Variable | ALK1 Mutation Carriers (n = 7) | BMPR2 Mutation Carriers (n = 18) | Mutation Noncarriers (n = 29) | All Patients (n = 54) | p Value |
|---|---|---|---|---|---|
| NYHA class I or II | 4 (57%) | 7 (39%) | 18 (62%) | 29 (54%) | |
| NYHA class III or IV | 0 | 3 (17%) | 5 (17%) | 8 (15%) | |
| Lung transplantation | 1 (14%) | 0 | 3 (10%) | 4 (7%) | |
| Death | 2 (29%) | 8 (44%) | 3 (10%) | 13 (24%) | |
| Rate of death (deaths/person-year) | 0.051 | 0.159 | 0.012 | 0.038 | 0.0002 |
| Characteristic | Hazard Ratio (95% Confidence Interval) | p Value |
|---|---|---|
| Gene mutation | ||
| ALK1 mutation vs mutation noncarriers | 5.14 (0.61–43.03) | 0.1205 |
| BMPR2 mutation vs mutation noncarriers | 12.54 (3.06–84.21) | 0.0003 |
| Gender (male vs female) | 2.62 (0.87–8.73) | 0.0869 |
Some BMPR2 and ALK1 mutations have been previously reported by other groups, and 7 BMPR2 mutation cases and 5 ALK1 mutation cases have been previously published by our colleagues ( Tables 6 and 7 ). Of 18 BMPR2 mutations, 6 were nonsense mutations, 3 were missense mutations, 2 were small deletions resulting in frame shifts, and 3 were splice-site mutations. MLPA analysis detected 4 exonic deletions ( Table 6 ). We identified 5 BMPR2 mutations that were not previously reported. All ALK1 mutations were missense mutations ( Table 7 ). One patient previously diagnosed with HHT subsequently developed features of PAH at the age of 11 years. We identified 2 ALK1 mutations that were not previously reported.
| Proband | Mutation Location | Mutation Category | Domain | Nucleotide Change | Amino Acid Change | Reference |
|---|---|---|---|---|---|---|
| 1 | Exon 1 | Nonsense | ECD | c.38 G>A | W13X | This study |
| 2 | Intron 2 | Splice-site | ECD | c.248-3 T>G | — | This study |
| 3 | Exon 3 | Nonsense | ECD | c.339 C>G | Y113X | Fujiwara et al |
| 4 | Exon 3 | Nonsense | ECD | c.339 C>G | Y113X | Fujiwara et al |
| 5 | Exon 3 | Missense | ECD | c.367 T>C | C123R | Machado et al |
| Fujiwara et al | ||||||
| 6 | Exon 3 | Deletion | ECD | c.247-? _420+? del | — | Machado et al |
| 7 | Exons 2-3 | Deletion | ECD | c.76-? _420+? del | — | Machado et al |
| 8 | Exons 2-3 | Deletion | ECD | c.76-? _420+? del | — | Machado et al |
| 9 | Intron 4 | Splice-site | ECD | c.529+2 T>C | — | This study |
| 10 | Exon 6 | Nonsense | KD | c.631 C>T | R211X | Thomson et al |
| 11 | Exon 6 | Missense | KD | c.727 G>C | E243Q | This study |
| 12 | Intron 8 | Splice-site | KD | c.1128+1 G>T | — | Pfarr et al |
| 13 | Exon 9 | Nonsense | KD | c.1207 C>T | Q403X | Uehera et al |
| Fujiwara et al | ||||||
| 14 | Exons 8-9 | Deletion | KD | c.967-? _1275+? del | — | Aldred et al |
| 15 | Exon 10 | Frameshift | KD | c.1376_1377 del GA | R459fsX10 | Sankelo et al |
| 16 | Exon 10 | Nonsense | KD | c.1397 G>A | W466X | Koehler et al |
| 17 | Exon 11 | Missense | KD | c.1472 G>A | R491Q | Deng et al |
| 18 | Exon 12 | Frameshift | CD | c.2289 del C | T762fsX9 | This study |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


