Other Neurohormonal Systems
Steven Goldsmith
Bradley A. Bart
Interference with the maladaptive consequences of imbalances in activity of the renin-angiotensin-aldosterone system (RAAS) and the adrenergic nervous system forms the bedrock of current therapy for left ventricular systolic dysfunction (LVSD) and the heart failure (HF) syndrome that frequently follows. Detailed discussion of the proof of this assertion, as well as an exploration of the potential mechanisms involved, are found elsewhere in this text. However, it is clear that the benefit of angiotensin-converting enzyme inhibitors (ACEIs), angiotensin receptor blockers, aldosterone antagonists, and beta-adrenergic blockers cannot be attributed primarily to direct effects on ventricular loading conditions or to a direct effect to improve myocardial function. Indeed, while such treatments stabilize and improve myocardial structure and function over time, they may have little or no acute beneficial effect; beta-blockers might even have adverse acute effects on such measures. These observations have forced us to search more deeply into the nature of the fundamental processes affecting progressive structural and functional abnormalities governing LV dysfunction (LVD) and HF. They have also made it considerably more difficult to design and conduct therapeutic trials, since clearly it is not always appropriate to use traditional functional measures as surrogates for assessing longterm effectiveness. In the case of certain vasodilators and inotropes, reliance on the response of traditional hemodynamic measurements as a guide to the design of long-term, placebo-controlled trials led to the unhappy result of achieving worse outcomes on active therapy.
We have therefore had to focus more closely on the impact of a potential new treatment, not just on ventricular function and loading conditions but also on the deeper biological processes suspected to be involved in HF (1). Unfortunately, these are not yet fully characterized. However, since treatments directed at neurohormonal imbalance have been our most successful efforts to date, it is logical to extrapolate from this success to other candidate pathogens within the neurohormonal milieu of HF. In general, most theoretically maladaptive elements of this milieu act on cell-surface receptors which are coupled to related intracellular signaling processes implicated in progressive structural and functional abnormalities. Hence, it is logical, based on the beneficial response to interfering with the receptors (or activity) of the RAAS and adrenergic nervous system, to extrapolate to interfering with the elements of other systems which may cause similar downstream effects over time. The two candidate substances most-studied to date are arginine vasopressin (AVP) and endothelin. Alternatively, since at least one system, involving the natriuretic peptide family, is most likely a beneficial counterresponse to LVD, vasoconstriction, and fluid retention, it has also been suggested that further increasing the level or effect of such peptides would be of benefit. We must stress that as of this writing there are not yet any approved agents for treating LVD or clinical HF based on antagonism of either AVP or endothelin, or on an attempt to chronically enhance natriuretic peptide activity. In fact, despite great theoretical promise and considerable effort, data regarding endothelin antagonism and natriuretic peptide enhancement are thus far disappointing and it is still too early to conclude anything definitive about AVP antagonists. Nonetheless, preliminary data with the latter approach are promising, and Phase III trials are under way. Because of both the clinical and pathophysiological importance of the attempt to further exploit neurohormonal imbalance as a target for drug development in HF, this chapter will attempt not only to summarize the rationale for and current status of
each of these three newer treatment strategies, but also to place the results to date in the overall context of HF therapy with an attempt to explicate the lessons learned thus far.
each of these three newer treatment strategies, but also to place the results to date in the overall context of HF therapy with an attempt to explicate the lessons learned thus far.
Vasopressin Antagonism
Rationale
The case for antagonizing the effects of AVP in HF rests on the possible contributions of AVP to both load-related and load-independent processes known to be important in progressive LVD (Fig. 12-1) (2). AVP is a nonapeptide made in the hypothalamus and stored in the posterior pituitary gland. Its secretion is primarily regulated by serum osmolal-ity but is also influenced by numerous hemodynamic and neurohormonal stimuli (3). AVP signals through at least three receptor subtypes: the V1a, V2 and V1b, or V3 receptors (4). These receptors are structurally dissimilar, are coupled to different intracellular processes, and are linked to very different end-organ responses. Briefly, the V1a receptor is G-protein-coupled, increases intracellular calcium via the IPS pathway, causes vasoconstriction in smooth muscle, and has both a positively inotropic and mitogenic effect in myocardial cells (5,6). The V2 receptor is also G-protein-coupled but acts via increasing cyclic adenosine monophos-phate (cAMP) to alter the expression of aquaporin channels in the renal collecting duct (7). The result is water retention (hence the other name for AVP, ADH—the antidiuretic hormone), V2 receptors also cause endothelium-dependent vasodilation but probably not at physiologic hormone levels, although this has only been investigated in normal humans (8). The V1b or V3 receptor is much less well-characterized, resides in the midbrain or hypothalamus, and is thought be involved in regulating cortisol secretion.
From the standpoint of LV dysfunction and HF, the V1a and V2 receptors are of the most importance. Given our current understanding of the pathophysiology of these syndromes, excessive AVP-mediated effects at either or both of these receptors could be maladaptive. Too much V1a signaling could contribute to increased systemic resistance, increased venous tone, and increased arterload and preload, thereby aggravating myocardial dysfunction and structural remodeling. Direct myocardial V1a effects could promote ventricular remodeling and hypertrophy independent of any load-related stimuli. An effective V1a antagonist might therefore be expected to be beneficial, both indirectly (via afterload and preload reduction) and directly (via interruption of adverse myocardial stimulation). Such effects would be analogous to those expected from interfering with excessive angiotensin II stimulation on the vasculature and myocardium.
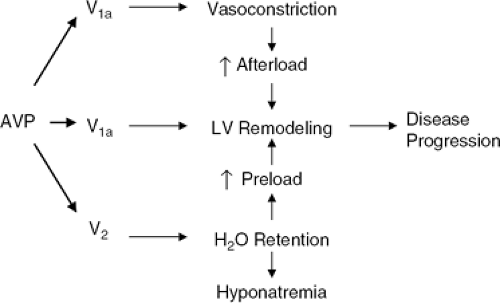 Figure 12-1 Proposed pathophysiological effects of arginine vasopressin (AVP) in congestive heart failure. By interacting with both V1a and V2 receptors, inappropriately increased AVP levels could adversely influence the progression of left ventricular dysfunction and remodeling in multiple ways. See the text for details. |
Excessive V2 stimulation could lead to volume expansion and an increase in both ventricular preload and afterload, again contributing to adverse LV remodeling and eventual contractile dysfunction. Hyponatremia is another possible result of excessive and inappropriate AVP secretion. Hyponatremia is emerging as one of the most powerful predictors of poor outcome in congestive heart failure (CHF), even in this era of modern therapeutics (9). While long-assumed to be just a marker for disease severity, this assumption is now under vigorous challenge, with a number of possible reasons being advanced for a possible bidi-rectional relationship between progressive CHF and a fall in serum sodium. Some of these hypotheses include possible undemse of ACEIs and diuretics because of fear of further lowering sodium; adverse effects of low sodium on quality of life and neurological function; and direct, adverse effects of low sodium on myocardial cell structure and function. If any or all of these prove true, chronically excessive V2 signaling could play a role in LVD and CHF well beyond the more obvious one of contributing to volume expansion. In this regard a most interesting recent clinical study demonstrated better outcomes in a group of patients with advanced HF as a function of directly increasing serum sodium, despite the challenging difficulties inherent in the use of hypertonic saline to achieve this goal (10).
Another related issue for considering a beneficial effect of interference with V2 signaling (if it is, in fact, excessive) would be the possibility of reducing dependence on loop diuretics as our primary means of maintaining volume homeostasis in CHF. Loop diuretics do effectively remove salt and water acutely, but their actual efficacy in either short- or long-term use has never been established. Their beneficial effects are rapidly attenuated, however, and there are a number of adverse results of their use which may either limit their effectiveness or, worse, actually contribute to disease progression and its consequences (11). These include electrolyte depletion, the myocardial and renal consequences of such depletion (particularly magnesium depletion and its effect on myocardial, vascular, and renal calcium overload), and direct stimulation of the very neurohormonal systems being targeted by other therapeutic agents. If volume control could be achieved, at least in part, without these effects, a safer and more effective diuresis could theoretically be the result.
The foregoing discussion makes it abundantly clear that there are many reasons to target AVP as a contributor to LVD and progressive CHF. The remaining part of the rationale rests on demonstrating excessive AVP signaling in the presence of these conditions. This demonstration must be based not just on hormone levels but also on proof that
interfering with AVP signaling has a meaningful and positive biological effect. Unfortunately, that is not always evident from acute experience and, in the absence of universally accepted surrogates, all acute experiences must be interpreted with caution. Nonetheless, there is abundant evidence from animal models for both excessive V1a and V2 signaling, some evidence (albeit scant) for excessive V1a signaling in human HF, and quite a bit of evidence for excessive, or at least demonstrable, V2 signaling in human HF.
interfering with AVP signaling has a meaningful and positive biological effect. Unfortunately, that is not always evident from acute experience and, in the absence of universally accepted surrogates, all acute experiences must be interpreted with caution. Nonetheless, there is abundant evidence from animal models for both excessive V1a and V2 signaling, some evidence (albeit scant) for excessive V1a signaling in human HF, and quite a bit of evidence for excessive, or at least demonstrable, V2 signaling in human HF.
Preclinical Studies
A possible role for AVP in the pathophysiology of HF has been explored in several animal models (12,13,14,15,16,17,18,19,20). In general, the results of these studies indicate the presence of significant V1a- and V2-mediated signaling. Most studies have focused on acute responses but several have looked at the impact of AVP antagonism over time.
Studies have been conducted with selective V1a antagonists in three separate models: adriamycin toxidty (13), tricuspid avulsion/pulmonary artery banding (12), and rapid ventricular pacing (14). All yielded positive results, using decreased systemic vascular resistance and/or improved cardiac output as the measure of V1a signaling. The combination of V1a antagonism and angiotensin II blockade produced greater improvement in myocyte function in an in vitro experiment than did either agent alone (15), but in vivo studies with pure V1a antagonism and other neurohormonal blockers have not been reported. There have been no studies reported with chronic administration of a V1a antagonist.
V2 antagonism has been studied in both the rapid ventricular pacing model (dogs) (14) and the post-myocardial infarction (MI) model (rats) (16). In each acute study, the administration of a selective V2 antagonist yielded a brisk diuresis (largely purely free water) and/or a decrease in cardiac filling pressure, depending on what was measured. A longer-term experience using a V2 antagonist in rats yielded a sustained response analogous to a clinical benefit (17).
Studies with combined V1a/V2 antagonism have also been performed. One acute study demonstrated apparent hemodynamic synergism with the administration of both a V1a and a V2 antagonist to dogs with pacing-induced failure (14). The V1a antagonist produced primarily an improvement in cardiac output with a fall in systemic resistance, while the V2 antagonist largely affected filling pressure. The combination, however, yielded greater and more sustained effects on each measure than did either alone. When an antagonist with both V1a and V2 antagonizing properties was administered in the same model, hemodynamic and renal responses were seen which were comparable to those observed with the administration of the individual agents (19). This agent (conivaptan, Astellas Pharma) has also been administered to rats with post-M1 LVD, both alone and in combination with an ACEI (20). That study focused on the impact of treatment on left and right ventricular remodeling. No hemodynamics beyond blood pressure were measured. At 1 week there was evidence of V1a signaling since blood pressure fell with conivaptan alone. Conivaptan alone did not exert major effects on either RV or LV mass, but the combination of conivaptan and captopril yielded greater effects on RV mass beyond that seen with captopril alone. There was a trend for a greater effect on the LV response.
The mechanism of the results is not clear but could involve interruption primarily of V2-mediated signaling, causing a reduction in preload, pulmonary venous pressure, and thus right ventricular remodeling. A role for the V1a moiety on venous tone, the pulmonary vasculature, or the LV could not be excluded, however. Perhaps the major importance of this study is the demonstration that AVP-mediated signaling is possible, and even more likely, in the presence of other clinically used neurohormonal antagonists. This is obviously relevant to the clinical situation and could be predicted by experimental work in models other than LVD or CHF which clearly established greater potency of AVP-mediated effects, especially V1a effects, in the presence of diminished activity of the RAAS and/or adrenergic nervous system (21,22).
Taken together, the preclinical results establish the presence of tonic V1a- and V2-mediated signaling in several animal models of ventricular dysfunction and HF, which can be interrupted by effective nonpeptide and peptide antagonists to AVP. However, only very limited information is available with chronic administration of any antagonist. What has been reported does suggest that it is possible to produce clinically relevant, sustained interference with either V2-mediated signaling alone or with both V1a and V2 signaling using a combined antagonist in association with interruption of the RAAS.
Clinical Studies
It has been known for some time that plasma AVP levels are elevated, or at least incompletely suppressed for the prevailing osmolality, in patients with chronic or acutely decompensated CHF (Fig. 12-2) (23,24,25,26). As with other measured neurohormones, AVP levels are correlated to poor outcome in this syndrome (27). AVP, similar to
plasma norepinephrine and atrial natriuretic peptide, is elevated early in the disease, including in patients with asymptomatic LVD (28). The reason for the increase in AVP secretion has thus far escaped clear delineation but must involve nonosmotic inputs (either hemodynamic or neurohormonal), since the osmotic regulation of the hormone is intact but shifted to higher plasma levels, with incomplete suppression in many patients despite very low serum sodium (29). The demonstration of either increased AVP levels or an association between plasma AVP and outcome in HF does not prove any pathophysiologicai contribution of AVP in HF, although such a relationship has been demonstrated with other neurohormones that have intra-cellular signaling pathways similar to AVP, such as norepinephrine, angiotensin II, and aldosterone. To do so, of course, requires proof that interfering with the secretion or effects of AVP produces chronic benefit in the syndrome of HF. Such data are not yet available for AVP but there are several acute and modestly prolonged experiences with AVP antagonists to consider.
plasma norepinephrine and atrial natriuretic peptide, is elevated early in the disease, including in patients with asymptomatic LVD (28). The reason for the increase in AVP secretion has thus far escaped clear delineation but must involve nonosmotic inputs (either hemodynamic or neurohormonal), since the osmotic regulation of the hormone is intact but shifted to higher plasma levels, with incomplete suppression in many patients despite very low serum sodium (29). The demonstration of either increased AVP levels or an association between plasma AVP and outcome in HF does not prove any pathophysiologicai contribution of AVP in HF, although such a relationship has been demonstrated with other neurohormones that have intra-cellular signaling pathways similar to AVP, such as norepinephrine, angiotensin II, and aldosterone. To do so, of course, requires proof that interfering with the secretion or effects of AVP produces chronic benefit in the syndrome of HF. Such data are not yet available for AVP but there are several acute and modestly prolonged experiences with AVP antagonists to consider.
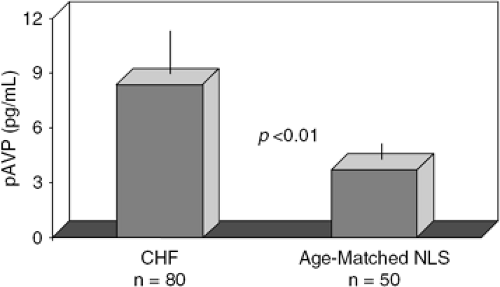 Figure 12-2 Plasma arginine vasopressin (AVP) levels from 80 patients with chronic congestive heart failure (CHF) and 50 age-matched control subjects. Plasma levels were collected on subjects between 1980 and 1985, prior to the widespread use of angiotensin-converting enzyme inhibitors (ACEIs). |
To date, human studies attempting to establish a role for AVP in the pathophysiology of HF at the plasma levels seen in the syndrome have included the infusion of exogenous vasopressin to determine its hemodynamic effects in patients with chronic HF; single-dose administration of a peptide antagonist to the V1a receptor in patients with HF and hypertension; administration of a V2 antagonist for up to 60 days in patients with chronic HF; and the acute administration of a mixed antagonist, also to patients with chronic HF.
Goldsmith et al. infused exogenous AVP into 11 patients with advanced, but stable, HF due to either ischemic or primary dilated cardiomyopathy (30). The result (Fig. 12-3) was a decline in stroke volume, an increase in systemic vascular resistance, and a rise in filling pressures at moderately increased plasma AVP levels. These findings suggested that modest increases in plasma AVP could produce adverse hemodynamic effects in chronic HF, presumably via the Via receptor.
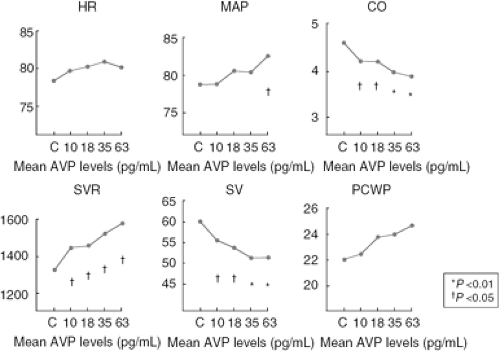 Figure 12-3 Hemodynamic effects of infusing exogenous AVP in patients with chronic CHF. Results were obtained via Swan-Ganz catheter in clinically stable patients. Goldsmith S, Francis GS, Cowley AW, et al. Hemodynamic effects of infused arginine vasopressin in congestive heart failure. J Am Coll Cardiol. 1986;8:779–783 .HR, heart rate; MAP, mean arterial pressure; SVR, systemic vascular resistance; CO, cardiac output; SV, stroke volume; PCWP, pulmonary capillary wedge pressure. |
Two single-dose studies using a selective V1a antagonist to AVP were reported at about the time of the infusion studies conducted by Goldsmith et al. Both studies found that if plasma AVP was increased, a fall in systemic vascular resistance and an increase in cardiac output occurred (Fig. 12-4) (31,32), These results thus complemented those by Goldsmith et al. and confirmed that hemodynamically important V1a-mediated signaling could be demonstrated under basal conditions in CHF if plasma AVP levels were increased.
A relevant, related study with the same compound in patients with severe hypertension demonstrated a fall in arterial pressure, even if plasma AVP levels were normal (33). This was an important observation since it established clinically relevant V1a signaling in humans with hypertension under basal conditions, and that (at least in this patient population) one could not necessarily predict a hemodynamic response to a V1a antagonist by measuring plasma AVP levels. No chronic experience in either HF or hypertension has been reported with selective V1a antagonism.
Selective V2 antagonism produces both an acute and sustained diuresis in patients with chronic CHF, which is largely an aquaresis since the major effect of these compounds is to increase free water clearance. One of these agents, tolvaptan (Otsuka Pharma), has now been given for 30 and 60 days to patients with CHF of mixed etiologies (34) and to those with acute decompensated heart failure (ADHF) due to LVD, respectively (35). The latter experience was placebo-controlled. Both studies confirmed sustained weight loss and a rise in serum sodium in those patients with hyponatremia (Figs. 12-5, 12-6). The drug was well-tolerated with only thirst as a common side effect. Neither study was powered for clinical endpoints as such, but the study in ADHF patients did include “worsening HF” as one measure, and it was unaffected. An intriguing post hoc analysis did demonstrate improved outcomes, including an unexpectedly large effect on mortality, in those patients with renal insufficiency or severe congestive symptoms. These results, taken together, demonstrate that one can produce a sustained interruption of V2 signaling in patients with chronic CHF using an appropriate agent
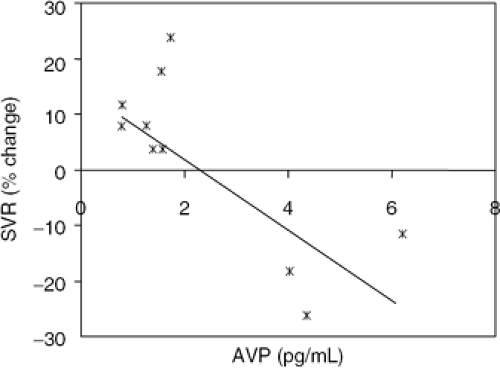 Figure 12-4 Effect of single-dose administration of a V1a antagonist on systemic vascular resistance (SVR) in patients with chronic, stable CHF. |
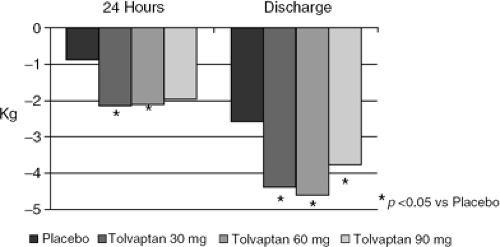 Figure 12-5 Effect of the V2 antagonist tolvaptan compared to standard care on body weight during hospitalization for acute decompensated heart failure. |
Only one experience has been published with a mixed V1a/V2 antagonist in patients with chronic CHF. This study, measuring the effect of various doses of conivaptan against placebo in patients with advanced but stable HF, demonstrated an acute fall in cardiac filling pressure and a brisk aquaresis, but no effect on systemic vascular resistance or stroke volume (36). Plasma AVP levels were low in this population of patients. The most likely explanation for these findings is an interruption primarily of V2-mediated signaling, but venodilating effects from the V1a-blocking moiety or a balanced effect of the agent on preload and afterload leaving stroke volume unchanged are not excluded. No chronic experience with this agent is available, although it has been given to many patients with hyponatremia, many of whom have CHF. These results suggest safety and a sustained effect on serum sodium for a period up to several days.
Implications of Current Data and Future Development Plans
As of this writing, no selective V1a antagonist is under active development for LVD or CHE The selective V2 antagonist tolvaptan is currently being investigated for short-term use against furosemide in chronic stable CHF, with the endpoints being renal function and plasma neurohormones. Tolvaptan is also being investigated against placebo in a 12-month study in chronic CHF with ventricular remodeling as the endpoint. Finally, this agent is being evaluated against placebo for its effect on mortality in a large population of patients with LVD, presenting with ADHF. Preliminary experience with the similar compound lixivaptan has been reported (37) and this agent may be slated for additional study, as well.
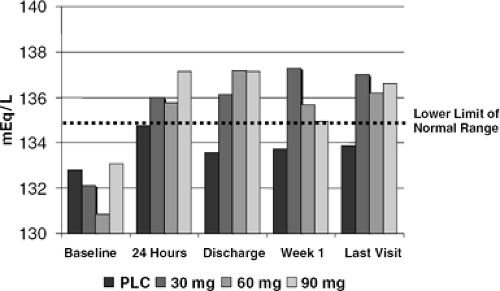 Figure 12-6 Effect of 60 days administration of the V2 antagonist tolvaptan on serum sodium in patients presenting initially with acute decompensated heart failure. |
The combined antagonist conivaptan has been studied in a Phase II pilot for acute decompensated HF; results are not yet available. A New Drug Application (NDA) has been submitted to the U.S. Food and Drug Administration (FDA) for approval of this agent specifically for hyponatremia. Due to interactions with the cytochrome P450 system, however, this agent will be used only in an intravenous form (if approved), limiting its potential use to acute or intermittent administration. As of this writing, to our knowledge no other mixed antagonist is being studied in CHF.
The case for interfering with AVP signaling is certainly strong based on our current pathophysiological concept of LV dysfunction and HF. There does seem to be active signaling at both the V1a and V2 receptors in the clinical syndrome, although at this point the evidence is stronger regarding the V2 receptor. However, it should be noted that there is much less information actually available with V1a or mixed antagonists, and that while both the preclinical and early clinical studies would predict a positive response to such agents, it may depend more on the nature of concomitant therapy or the prevailing plasma AVP level than does the V2 signaling. In any event if one considers the possible utility of vasopressin antagonism in CHF, several important points should be bome in mind.
A pure V1a antagonist would have the expected hemodynamic and myocardial effects but could, if AW levels rose in response to competitive displacement of hormone from V1a receptors, lead to unwanted water retention. An increase in ventricular preload might therefore offset any gains from lower afterload or direct benefits to the myocardium. A pure V2 antagonist may well lead to sustained water diuresis but as AVP levels rise in response to increased plasma osmolality and/or competitive displacement of hormone from V2 receptors, undesirable V1a stimulation could occur. For these reasons, it might be most useful to employ agents that block both receptor sites, although the impact of the V1a antagonizing moiety might not be readily apparent in shorter-term studies and the utility of V2 receptor antagonism in patients not already volume-expanded might well be questionable. Since it may be possible to adjust the relative degree of receptor antagonism in newer compounds, the use of a particular agent might be tailored to particular clinical settings. For acutely ill or chronically congested patients, a major component of V2 antagonism would be desirable, while for prophylactic use in patients with LVD but without a great deal of volume expansion, an agent with primarily V1a activity might be best.
The settings in which these agents might be used also deserve consideration. Chronic, stable LVD with or without mild CHF now carries a very favorable prognosis (38). The success of current therapies renders it challenging from a statistical point of view to demonstrate the effectiveness of
newer treatment. On the other hand, episodes of ADHF and the presence of persistent congestion identify a high-risk population (39) in which it may be easier to demonstrate effectiveness of newer treatment. Effective V2 antagonism, especially if combined (for the reason previously discussed) with V1a antagonism, could be very useful in these patients since the most obvious and direct effect of these agents is volume removal. Given the limitations and toxicity of diuresis produced only by loop diuretics, adjunctive or replacement diuretic therapy with a V2 antagonist could provide more effective (and actually safer) long-term volume control.
newer treatment. On the other hand, episodes of ADHF and the presence of persistent congestion identify a high-risk population (39) in which it may be easier to demonstrate effectiveness of newer treatment. Effective V2 antagonism, especially if combined (for the reason previously discussed) with V1a antagonism, could be very useful in these patients since the most obvious and direct effect of these agents is volume removal. Given the limitations and toxicity of diuresis produced only by loop diuretics, adjunctive or replacement diuretic therapy with a V2 antagonist could provide more effective (and actually safer) long-term volume control.
Finally, hyponatremia is, as previously noted, a marker for very poor outcome in CHF. Agents such as V2 antagonists that rapidly and safely correct hyponatremia conceivably could lead directly to improved outcomes in this group of patients if the association is causative in anyway. Since V2 antagonists have been shown to correct and maintain sodium homeostasis over a period of weeks in patients with CHF and hyponatremia, longer-term study of V2 or mixed antagonists specifically in this patient group seems warranted.
Additional Neurohormonal Targets in Heart Failure: Endothelin and Neutral Endopeptidase
As discussed in the introduction to this chapter, a number of additional neurohormonal targets for the treatment of HF have been suggested, based on the success of agents which interfere with the adrenergic nervous system and the RAAS. This section of the chapter will review the rationale and experience for targeting endothelin and neutral endopeptldase.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


