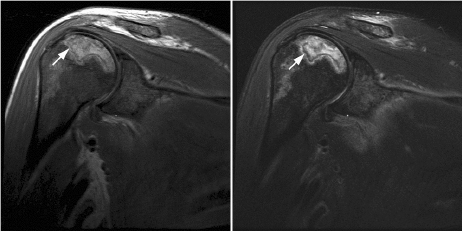
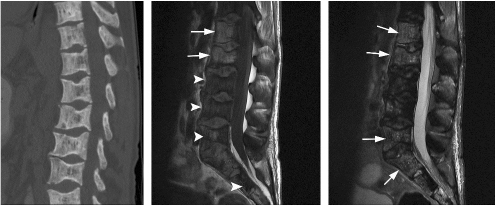
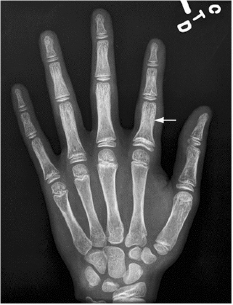
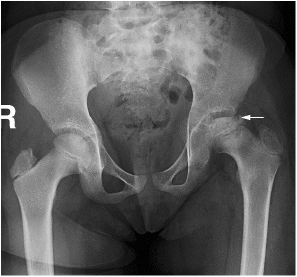
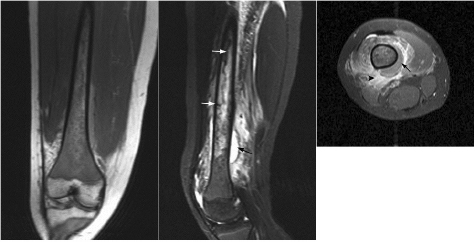
CHAPTER 8 The “Lazarus Dilemma”: It’s not the death that gets you. It’s the resurrection! Dr. Henry J. Mankin Bone infarction occurs when vascular flow is sufficiently impaired, or blocked, causing osteonecrosis. About 10,000–20,000 new cases are estimated each year; 21–37% of which are secondary to corticosteroid use.19, 26 In the process of avascular necrosis, initially a variety of immunologic cells with repair capabilities from surrounding tissues, and vasoactive cytokines infiltrate the area, causing acute inflammation. Over the subsequent weeks and months, bone-resorbing osteoclasts in an acid-rich microenvironment, slowly bore into, or erode, the necrotic bone. These reparative replacement processes slowly begin at the interface between the viable and infarcted (non-viable) bone; however, these are often too slow to maintain the mechanical integrity of the periarticular bone, supporting the articular cartilage, leading to collapse of the subchondral supporting bony architecture, and ultimately, articular destruction. Without the underlying structure or foundation of subchondral bone, the remaining tissue architecture collapses, as is observed with avascular necrosis in various disorders to be discussed, including sickle cell anemia and Gaucher disease. The actual process and cellular mechanism of corticosteroid-induced avascular necrosis is poorly understood. However, when present, it often produces significant morbidity and attendant disabilities, requiring various surgical management options, most typically joint replacement.19 The process in diaphyseal bone is similarly endosteal, affecting the cancellous or spongiosa bone marrow bearing areas, but typically manifests as an acute “bone crisis.” The resorption or restoration process in the diaphysis typically is less critical, as cortical bone is relatively difficult to remove and naturally strong, as opposed to epiphyseal or metaphyseal, subchondral loci. Associated co-morbidities, such as osteomyelitis in sickle cell infarcts, are problematic, but can be effectively minimized with prophylactic antibiotic use, particular in infants and young children.7, 9 A similar issue arises in mandibular infarctions associated with osteoclast-inhibiting medications, such as bisphosphonates, as used in the treatment of osteoporosis, typically in locations juxtaposed to dental extraction sites. Sickle cell disease (SCD) is caused by a selective genetic mutation in the beta chain of hemoglobin leading to the substitution of valine for glutamic acid at the sixth amino acid. This substitution results in an abnormal hemoglobin that polymerizes in physiologic states such as hypoxia or dehydration, leading to a distortion in the shape of red blood cells and giving them their characteristic sickled appearance.This gene is widely understood to have evolved and been preserved due to its protective effects against malaria when inherited as a single gene, and is observed in African, Middle Eastern, Indian, and Mediterranean populations. The autosomal recessive sickle cell gene is present in 8% of the black African American population. While roughly 2 million people have sickle cell trait, one in every 500 black African Americans suffer from overt sickle cell disease, which currently affects approximately 70,000 people in the United States.1 In addition to hemoglobin S, other hemoglobin variants can be co-inherited in a double heterozygote fashion and can also lead to human disease. Most notably, hemoglobin C in which glutamic acid at the sixth residue on the beta chain is replaced by lysine causes symptoms when co-inherited with hemoglobin S that are typically milder than those found in patients homozygous for hemoglobin S. An important exception is that patients with SC disease have more bone complications than patients with sickle cell disease. Sickled hemoglobin leads to red cell hemolysis and a shortened red cell life span. Sickling also results in increased adhesion between cells, ultimately leading to vasoocclusive events in various organs including bone, brain, kidney, retina, liver and spleen.5 Vasoocclusive crises can be precipitated by a low oxygen tension environment, which triggers hemoglobin precipitation leading to the sickling process. Sickling ultimately leads to thrombosis of the terminal vasculature.6 Due to relatively low blood flow rates in bone; the osseous architecture creates a pathophysiologic niche for sickling to occur. In addition to in situ thrombosis, hemolysis leads to nitric oxide depletion due to scavenging by serum-free hemoglobin and leads to depletion of arginine, which is necessary for nitric acid production.15 The combination of arterial constriction due to reduced nitric oxide and thrombosis leads to the clinical features seen during a vasoocclusive crisis. During a typical vasoocclusive crisis, patients experience pain in long bones, back, abdomen, and chest. Additionally, patients can have pulmonary complications of a sickle cell crisis and develop an acute chest syndrome characterized by fever, hypoxia, chest pain, and shortness of breath. SCD often presents with characteristic osseous imaging features. Increased hemolysis results in persistence of the appendicular red marrow with widening of the medullary spaces and thinning of the cortical bone. In the spine, cortical thinning produces a biconcave deformity of the vertebral bodies (Fig. 8.1). Vasoocclusive events can lead to osseous infarction, typically occurring in the medullary cavity or epiphyses.6 On MR imaging, chronic medullary infarcts have a thin low signal rim on all imaging sequences with a variable signal of the center. Osseous infarction in young children can occur in the small tubular bones of the hands and feet, termed dactylitis.25 Radiographs demonstrate patchy areas of lucency with periosteal new bone formation and related growth abnormalities (Fig. 8.2). Fig. 8.1. Images of the lumbar spine in a patient with sickle cell anemia. (A –left): Sagittal CT image shows diffuse sclerosis and coarsening of the trabeculae and central endplate compression deformities resulting in characteristic H-shaped vertebral bodies. (B –middle): Sagittal T1-weighted MR image demonstrates diffuse low signal intensity of the vertebral bodies consistent with hyperplastic hematopoietic red marrow (arrow-heads), which is replacing the normal bright fatty marrow (arrows). Concavity of the vertebral endplates at multiple levels is also seen. (C –right): Sagittal fat-suppressed T2-weighted image shows areas of high signal, consistent with active infarction (arrows). Epiphyseal infarction (also commonly referred to as osteonecrosis, or avascular necrosis) often presents in the femoral and humeral head. On T2-weighted MR imaging, high signal intensity indicative of bone marrow edema and a serpiginous double line that consists of a hyperintense inner border and hypointense periphery can be seen (Fig. 8.3).8 In later stages of avascular necrosis, radiographs typically demonstrate crescent-shaped subchondral lucencies and articular surface collapse (Fig. 8.4).17 In addition to osteonecrosis, osteomyelitis is seen in patients with sickle cell disease. Because patients homozygous for the sickle gene typically autoinfarct their spleens early in life, there is a high incidence of infection due to encapsulated organisms in patients with sickle cell anemia.7, 9 Interestingly, patients with sickle cell anemia are also prone to osteomyelitis with gram negative organisms such as salmonella. Distinguishing acute infection from infarction by imaging alone can be difficult but is important in light of correct and timely treatment. Radiographic features are often non-specific, showing periosteal inflammation, osteopenia, and sclerosis. Ultrasonography and radioisotope bone imaging may provide useful information in the early stages of acute osteomyelitis by showing features typical of inflammation.11 MR imaging, in particular contrast enhanced T1-weighted imaging, is valuable by showing inflammatory osseous changes and associated soft tissue abnormalities in the setting of infection (Fig. 8.5).13, 27 Fig. 8.2. Frontal radiograph of the left hand in a child with sickle cell anemia shows shortening of the second proximal phalanx (arrow). The restricted growth is secondary to childhood dactylitis and cartilage infarction during the growth period. Fig. 8.3. MR images of the right shoulder in a patient with sickle cell anemia. Coronal oblique proton density-weighted (A –left) and fat-suppressed T2-weighted (B –right) images demonstrate abnormal bone marrow signal and edema within the epiphysis with serpiginous linear signal abnormality (arrows), consistent with avascular necrosis. Fig. 8.4. Pelvic radiograph in a patient with sickle cell anemia shows sclerosis and crescent-shaped subchondral lucencies with articular surface collapse of the left femoral capital epiphysis (arrow), consistent with osteonecrosis. The most commonly seen bone complication in patients with sickle cell anemia is osteonecrosis of the humeral or femoral head, which is also called avascular necrosis (AVN). AVN likely results from infarction of the tenuous vascular supply to the femoral or humeral head but vitamin D deficiency has also been identified as a contributing factor.2 Treatment for AVN is not standardized. Total hip arthroplasty can provide pain relief, and restoration of function in selected symptomatic patients with sickle cell disease and AVN.14 Asymptomatic disease may not require therapy.20 A recent Cochrane analysis was only able to identify one randomized trial involving hip AVN in patients with sickle cell disease. That study concluded that there was no advantage to adding hip core decompression to standard physical therapy with respect to major complications and pain.18 Additionally, cementless prosthetic components may provide some advantage over those that are cemented.12 For osteonecrosis of the humeral head, there is consensus that if left untreated, humeral head collapse will result, necessitating surgical intervention.22 As with hip arthroplasty, the optimal method of repair has not been determined. Fig. 8.5. MR images of the femur in a patient with sickle cell anemia and acute fever, lower extremity pain, and swelling. Coronal T1-weighted image (A –left) demonstrates diffuse low signal replacement of fatty marrow due to red marrow reconversion. Sagittal fat-suppressed T2-weighted image (B –middle) shows increased T2 signal involving the entire diaphysis with serpiginous areas of low signal intensity (white arrows), consistent with medullary bone infarcts. A T2 hyperintense collection around the lower aspect of the right femoral shaft (black arrow) is noted. Axial fat-suppressed T1-weighted image with intravenous gadolinium (C –right) demonstrates a non-enhancing collection (arrow) with surrounding enhancing inflammatory changes (arrowhead) which was found to represent an abscess likely related to underlying osteomyelitis.
OSTEONECROSIS IN PATIENTS
WITH SICKLE CELL ANEMIA
AND OTHER HEMATOLOGIC DISORDERS
8.1 Introduction
8.2 Sickle Cell Disease and Osteonecrosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


