Loop diuretics
Hypersensitivity reactions
Extracellular fluid volume depletion
Hypokalemic alkalosis
Hypomagnesemia∗
Ototoxicity
Distal convoluted tubule diuretics
Hypersensitivity reactions
Hyponatremia
Hypokalemic alkalosis
hyperglycemia/diabetes
hyperuricemia/gout
Hypomagnesemia
Hypokalemia and prerenal azotemia, when combined with loop diuretics
Potassium sparing diuretics
Hypersensitivity
Hyperkalemia
Metabolic acidosis
Azotemia
Gynecomastia, vaginal bleeding (spironolactone)
Classification and Mechanisms of Action
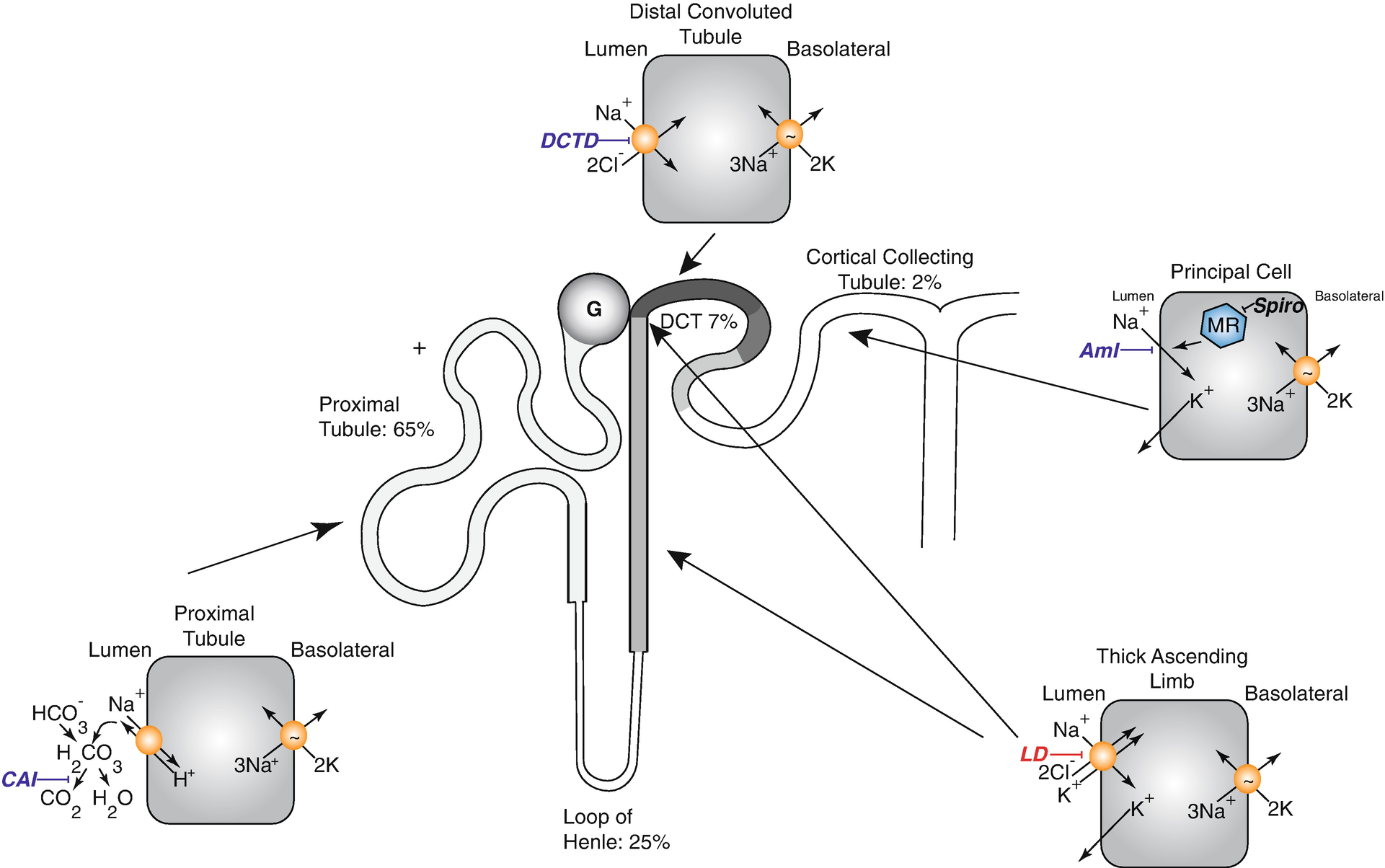
Diagram of nephron showing sites of sodium reabsorption and diuretic action along the nephron. CAI carbonic anhydrase inhibitors, DCTD distal convoluted tubule diuretic, Aml amiloride (also triamterene), LD loop diuretics, MR mineralocorticoid receptors in the CD are blocked by spironolactone and eplerenone action (note that these steroid antagonists act within the cell). Percentages show approximate percentage of sodium reabsorption by associated segment. The translocation pocket for ions and diuretic binding of the Na-K-2Cl cotransporter is shared [73, 74]
There are 2 major classes of potassium-sparing diuretics, which act along the aldosterone-sensitive distal nephron. The first includes drugs that block apical sodium channels (amiloride and triamterene), whereas the second includes drugs that antagonize mineralocorticoid receptors (spironolactone, eplerenone). A new non-steroidal mineralocorticoid blocker, finerenone, which is structurally unrelated to the others, is currently in phase III clinical trials. The mineralocorticoid blockers act within cells and do not require secretion into the tubular lumen.
Bioavailability of Diuretics Determining the Dose and Frequency of Administration
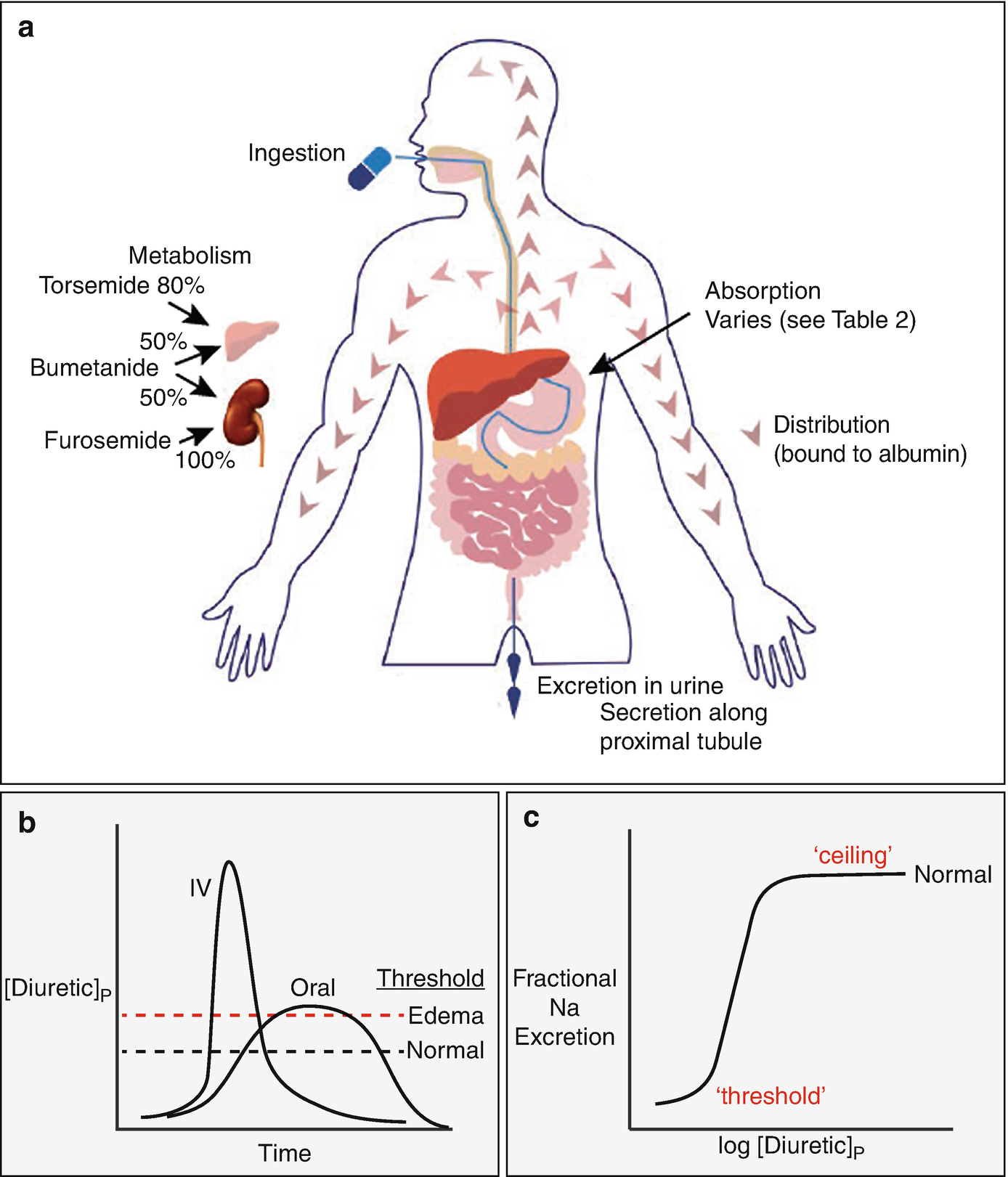
(a) shows features of absorption, distribution, metabolism, and excretion (so-called ADME of drugs). (b) compares the plasma diuretic concentration as a function of time following oral or intravenous diuretic administration. The dashed lines show natriuretic thresholds in normal individuals and in those with edema. Note that the primary determinant of natriuresis is the time above the threshold, indicating why route of administration has different effects in stable patients and in those with severe edema. In a normal individual, an oral dose may be effective, whereas it may not be in edema, despite retained bioavailability. (c) Classic dose response curve, plotted versus the logarithm of the plasma concentration. Note the threshold for natriuresis and the maximal level, often called the ceiling. (Figure adapted from Ref. [74])
Pharmacokinetics of commonly used diuretics
Diuretic | Oral bioavailability, % | Elimination half-life, hours | |||
|---|---|---|---|---|---|
Normal | CKD | Cirrhotic ascites | Heart failure | ||
Furosemide | 50 (10–100) | 1.5–2 | 2.8 | 2.5 | 2.7 |
Bumetanide | 80–100 | 1 | 1.6 | 2.3 | 1.3 |
Torsemide | 68–100 | 3–4 | 4–5 | 8 | 6 |
Hydrochlorothiazide | 55–77 | 6–15 | Prolonged | ||
Chlorthalidone | 61–72 | 40–60 | Prolonged | ||
Metolazone | 70–90a | 14–20 | Prolonged | ||
Amiloride | ~50& | 6–26 | 100 | Not changed | |
Spironolactone | >90 | 1.5∗ | ∗∗ | ||
Based on oral bioavailability, when a patient is switched from an intravenous to oral loop diuretic, the dose of bumetanide or torsemide should be maintained, whereas the dose of furosemide should be doubled [5]; in practice, however, and as discussed further below, other factors affect diuretic efficacy and a fixed intravenous/oral conversion cannot be given [6]. Gastrointestinal absorption of thiazide and other potassium sparing diuretics is fairly rapid and predictable, and oral bioavailability ranges from 65% to 90%.
The loop diuretics have steep dose response curves. This property is often neglected in clinical practice but is crucial to optimal use. Figure 5.2c shows a a schematic of a typical natriuretic response plotted versus the logarithm of the plasma diuretic concentration. Inspection reveals that there is little diuretic or natriuretic effect when the plasma concentration is low. Once the concentration exceeds a certain level, often called the diuretic threshold, the response increases, and even small further increases caused substantially increased sodium excretion. Although such relations are typically plotted as the logarithm of the diuretic concentration or dose, clinicians do not typically think in logarithmic terms. This underlies the reasoning behind the common recommendation to double the dose, if no response is obtained to the first dose. Although the dose response looks steep, when plotted logarithmically, it is less so when plotted in a linear manner, and a small increase will often be ineffective. At higher concentrations, a plateau or ceiling is reached, with progressively higher plasma concentrations failing to elicit more natriuresis. Although this fact has been used to invoke the concept of ceiling doses of loop diuretics, we will argue that increasing a diuretic dose above this ceiling often elicits more natriuresis, owing to pharmacokinetic considerations (see below).
As should be evident from these plots, a diuretic dose must exceed the threshold to be effective; yet the failure to give a dose that exceeds the threshold is one of the most common errors in diuretic usage. The problem is that the threshold is not easily estimated in an individual, especially an individual with cardiorenal syndrome. While nearly all healthy individuals will respond to 20 mg of oral furosemide (or its equivalent), patients with conditions that predispose to ECF volume expansion and edema need higher doses, since these conditions alter both the pharmacokinetics and pharmacodynamics of diuretics as discussed below. It is little wonder that an empirically selected dose may be ineffective. Below, we will provide broad generalizations about dose adjustments for individuals within a variety of settings. Yet adherence to algorithms may lead to diuretic failure. Instead, it is often best to approach a patient as an n of 1 trial. Start with a dose consistent with the clinical guidelines (more aggressive for acute edema, more conservative for more chronic processes) and then adjust the dose according to the response.
Although low bioavailability is a concern with furosemide, a larger problem may be its inconsistent bioavailability. Furosemide absorption varies from day to day in an individual, and between individuals [7, 8]. Absorption is also affected by food consumption, unlike that of bumetanide or torsemide [9, 10], although the clinical significance of this effect has been doubted [1]. The more consistent bioavailability of torsemide, compared with furosemide, and its relatively longer half-life, have suggested that it may be a superior loop diuretic, as suggested by 2 small clinical trials [11–14]. A recent non-randomized post-hoc analysis of the large Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure (ASCEND-HF) has suggested that patients with heart failure discharged on torsemide have lower mortality [15]. In contrast, the bioequivalent doses of 2 loop diuretics given in a double-blind randomized crossover trial, failed to demonstrate superiority of torsemide with respect to natriuresis or 24-h ambulatory blood pressure control in chronic kidney disease (CKD) patients [16]. The compelling differences in pharmacokinetics suggest that torsemide might be superior clinically, but this needs to be tested in sufficiently powered and rigorous trials. ToRsemide compArisoN With furoSemide FORManagement of Heart Failure (TRANSFORM-HF) is a large-scale, pragmatic, randomized, unblinded clinical effectiveness study comparing torsemide versus furosemide as treatment for heart failure. The study is sponsored by National Heart, Lung, and Blood Institute (NHLBI), actively recruiting patients and expected to be complete by 2022 (NCT 03296813).
Gastrointestinal absorption can be slowed, especially during exacerbations of heart failure, although again, this problem is worst with furosemide [17]. Even though total bioavailability is typically maintained in these situations, natriuresis may be impaired when absorption is slowed, especially given a concomitant increase in natriuretic threshold, as diagramed in Fig. 5.2b. As an example, the areas under the curves for arbitrary intravenous and 2× oral furosemide doses may be similar, but the differences in shapes of those curves may lead to a diuretic being effective when given intravenously, but not orally. As the diuretic threshold is higher in cardiorenal syndrome, this difference may be especially relevant in this situation. This is likely to explain the common observation that intravenous doses of loop diuretics, which achieve higher peak levels, may be effective when oral doses lose their efficacy, especially if the natriuretic threshold is increased. Not surprisingly, heart failure exacerbation requiring IV diuretics remains one of the major reasons for visits to emergency department and outpatient clinics, and for hospital admissions. In this context, development of subcutaneous (SC) furosemide has garnered increasing optimism as a safe and effective outpatient alternative to the traditional hospital-based IV diuretic strategy. Recently, 2 proof-of-concept studies have demonstrated similar urine output with a pH-neutral formulation of SC furosemide compared to IV furosemide in outpatients presenting with decompensated heart failure [18]; and longer duration at therapeutic plasma levels and more urine output compared to oral furosemide [19]. Subcutaneous administration of buffered furosemide was well tolerated with no evidence of any drug-induced skin reactions. The possibility of delivering an IV equivalent diuretic agent at home, if proved efficacious in larger trials, could be transformative for HF care delivery.
Volumes of Distribution, Metabolism and Half-lives
Loop diuretics are organic anions that circulate tightly bound to albumin (>95%). Thus, their volumes of distribution are low, except during extreme hypoalbuminemia [20]. This has suggested that severe hypoalbuminemia might impair diuretic effectiveness, owing to impaired delivery to the kidney, and that albumin administration might enhance natriuresis. This conjecture was supported in an early proof-of-concept study [20], but subsequent larger studies have produced mixed results. A relatively recent meta-analysis concluded that the existing data, albeit of poor quality, suggest transient effects of modest clinical significance for co-administration of albumin with furosemide in hypoalbuminemic patients [21]. One concern about the more recent studies, many of which have been negative, is that most have only enrolled patients whose serum albumin concentrations exceeded 2 g/dL. Owing to physiological plausibility and anecdotal experience, most experts would still consider adding albumin infusion for refractory patients who are severely hypoalbuminemic. Yet, extreme caution should be used when treating patients with cardiorenal syndrome, as their extracellular fluid volume is typically expanded substantially; as albumin infusions expand the extracellular fluid volume, they should be avoided in most patients with cardiorenal syndrome.
Approximately 50% of an administered furosemide dose is excreted unchanged into the urine. The remainder appears to be eliminated by glucuronidation, predominantly also in the kidney. Torsemide and bumetanide are eliminated both by hepatic processes and urinary excretion, although hepatic metabolism may predominate, especially for torsemide [22]. The differences in metabolic fate mean that the half-life of furosemide is prolonged in kidney failure, where both excretion by the kidney and kidney-mediated glucuronidation are slowed. In contrast, the half-lives of torsemide and bumetanide tend to be preserved in patients with kidney dysfunction [23]. While the ratio of equipotent doses of furosemide to bumetanide is 40:1 in normal individuals, that ratio declines as kidney disfunction worsens [24]. Although this apparent increase in furosemide potency may seem beneficial, it also likely increases the toxic potential of furosemide when it is used in very high doses. Deafness and tinnitus from loop diuretics appear to result primarily from high serum concentrations, which inhibit a Na-K-2Cl isoform (NKCC1, encoded by SLC12A2). This transport protein, which is different from that reabsorbs salt in the kidney, is expressed by the stria vascularis and participates in secretion of K+-rich endolymph [25, 26]. Although this complication was seen more frequently in the past when very large bolus doses of loop diuretics were employed to forestall dialysis [27], one relatively recent meta-analysis of furosemide use for patients with acute kidney injury, suggested that the odds ratio for hearing loss was >3 when high dose furosemide was used; it should be noted, however, that the doses cited in that analysis (1–3 grams daily) exceeded those currently recommended [28]. The tendency of bolus infusion to lead to high peak furosemide concentrations is one reason that many investigators recommend continuous infusions instead [29].
Although loop diuretics are small molecules, they typically undergo little glomerular filtration. As they exert their actions by binding to transport proteins along the luminal membrane of thick ascending limb cells, to gain access to the tubular fluid and therefore to their sites of activity, they must be secreted. This is likely true for all three loop diuretics, although some data suggest that bumetanide is also delivered into the tubule lumen by filtration [30]. However, most evidence still suggests that bumetanide gains entry primarily via secretion [31]. Peritubular uptake by cells along the proximal tubule is mediated by the organic anion transporters OAT1 and OAT3, whereas the apically located multidrug resistance-associated protein 4 (Mrp-4) appears to mediate at least a portion of secretion into the tubular fluid. Mice lacking OAT1, OAT3 or Mrp-4 are resistant to loop and thiazide diuretics illustrating the functional importance of diuretic secretion for diuretic effectiveness [30, 32].
Impact of Drugs and Chronic Kidney Disease on the Effectiveness of Diuretics
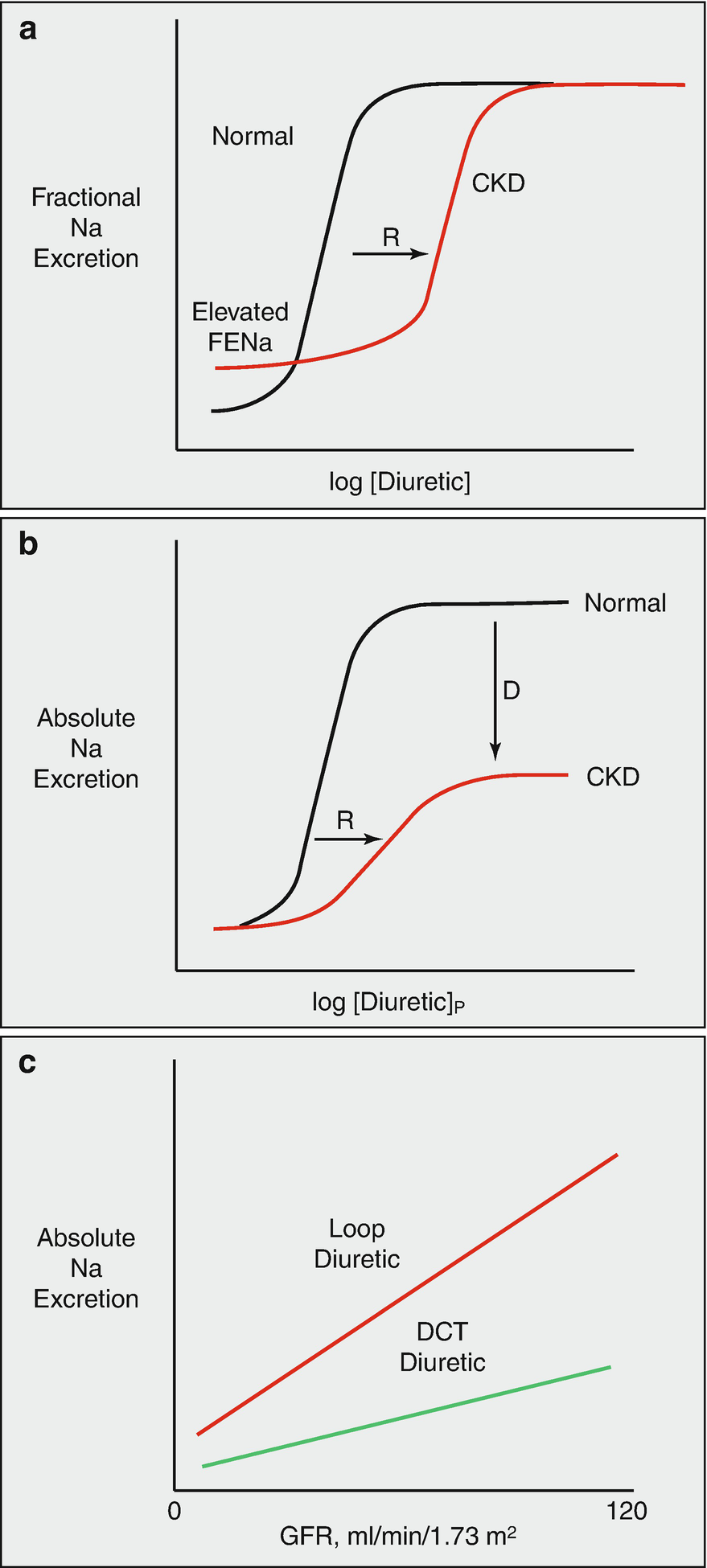
(a) effects of chronic kidney disease on diuretic actions. Note that, in CKD, baseline fractional sodium excretion is high, to maintain absolute rates of sodium excretion equal to intake. There is a shift in the dose response curve to the right (R), primarily owing to impaired diuretic secretion, but no change in the ceiling effect. (b) shows the same relationship plotted versus absolute rates of sodium excretion. The same rightward shift is evident, but the ceiling is lower, owing to the GFR reduction. (c) compares effects of loop diuretics and distal convoluted tubule diuretics on absolute sodium excretion, given a retained effect on fractional excretion. (Figure adapted from Ref. [74])
There are additional reasons that patients with poor kidney function are resistant to loop diuretics. Metabolic acidosis, which is frequently observed in uremia, depolarizes the membrane potential of proximal tubule cells [36] which also decreases organic anion secretion, an effect that may explain why diuretic secretion is enhanced by alkalosis [37]. In addition to a shift in the dose-response curve, patients with poor kidney function and those taking non-steroidal anti-inflammatory drugs (NSAIDs) have a downward shift of the ceiling natriuresis, when expressed as absolute sodium excretion (rather than fractional). The mechanism for resistance attributable to NSAIDs is complex. Loop diuretic inhibition of NaCl reabsorption at the macula densa stimulates both renin secretion and prostaglandin production, the latter predominantly via cyclooxygenase-2 (COX-2) [38]. When this happens, prostaglandin E2 feeds back on tubules, contributing to the resulting natriuresis by inhibiting NaCl transport along the thick ascending limb and collecting duct [39, 40]. NSAIDs block this prostaglandin-mediated natriuresis. When used chronically, NSAIDs increase the abundance and activity of NKCC2 along the thick ascending limb [41]. Additionally, loop diuretics inhibit the second transporter isoform, NKCC1, mentioned above, which, in addition to being expressed in the ear, is also expressed by vascular smooth muscle cells; loop diuretics contribute to vasodilation of the glomerular afferent arteriole by blocking this transporter [42], thus helping to maintain glomerular filtration rate despite a lower ECF volume. Again, this compensatory adaptation is largely dependent on prostaglandin production and can be blocked by NSAIDs. The clinical impact of these effects is evident in the association between recent use of NSAIDs and risk for hospitalization in patients with heart failure [33]. In fact, in the setting of loop diuretics and angiotensin-converting enzyme inhibitors or angiotensin receptor blockers, the addition of a third class of drug that alters intrarenal hemodynamics such as NSAIDs, is associated with acute kidney injury [43].
Intrinsic kidney dysfunction also impairs the natriuretic response to diuretics through a different mechanism (Fig. 5.3b). It is frequently noted that the maximal natriuretic capacity of loop diuretics is maintained in the setting of chronic kidney disease, when natriuresis is measured as a fraction of filtered load (FENa). Yet the maximal natriuretic effect of these diuretics, when measured as the more clinically relevant absolute rate, is markedly reduced. This is because, as glomerular filtration rate and filtered sodium load decrease, kidneys suppress sodium reabsorption by the remaining tubules to keep sodium excretion equal to dietary salt intake. This suppression occurs in part along the thick ascending limb, so that even when a diuretic reaches the segment and inhibits the transporter, its net effect is reduced. Thus, NSAIDs and CKD cause diuretic resistance both by shifting the diuretic dose response curve to the right (which can be overcome by higher doses) and by reducing maximal natriuresis (which cannot overcome by higher doses, compare Fig. 5.3a, b).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


