Fig. 7.1
The canonical pathway of microRNA processing (Reprinted by permission from Macmillan Publishers Ltd: Winter et al. [97] Nature Publishing Group)
Computational Biology, Our Venue to Understand MicroRNA Network Biology
As mentioned the complexity of microRNA regulations is derived from the fact that a microRNA can target multiple mRNAs and multiple microRNAs can target a single mRNA. Thus approaches that integrate signaling from microRNAs, to mRNAs and proteins are required. However, since the microRNA field is relatively young, classification and nomenclature of microRNAs, and databases of their predicted and validated targets were required.
An online microRNA database, named miRBase, has a list of all microRNAs that are currently identified in all organisms including humans [26]. The nomenclature of microRNAs has also been standardized [26–28]. One of the more basic computational methods to identify microRNA targets and potential function includes a simple sequence analysis [29]. Such out-of-context base sequence analysis predictability has a high rate of false positive and false negative results, which makes such analyses challenging and usually require biological validation and direct demonstration of effect on target genes.
MicroRNAs are frequently co-expressed with host genes, as well as with neighboring microRNAs [30], which highlights the networking effect and the ‘team-spirit’ of microRNAs, working in consortiums, rather than individually or independent of other transcription factors. The multiplicity of targets of a single microRNA [5] logarithmically increases the effect of any aberrancy in microRNAs on multiple pathways within a specific disease and potentially multiple simultaneous disease states. Efforts at creating dynamic regulatory network analysis capability and creating probabilistic modeling methods are promising [31]. Previously unknown microRNAs and new roles of known microRNAs could be predicted and identified using probabilistic modeling methods [such as the MiRNA Dynamic Regulatory Events Miner (mirDREM)] that use models to reconstruct a dynamic virtual network that explains joint regulation of microRNAs and other transcription factors of expression of protein-coding genes over a period of time [31]. Once these microRNAs and/or their roles are identified, they would then be validated/confirmed using proliferation assays for example. Transcription factors and microRNAs regulate each other, and in combination regulate protein-coding genes under different disease states [32, 33]. Computational transcription factor-microRNA networks are available for some genomes and diseases [14, 34, 35]. A wealth of data has been generated by the ENCODE project [36], however integrated analysis and experimental verification of these interactions are still ongoing.
Clinical Phenotype of Pulmonary Arterial Hypertension
As detailed in the other chapters of this book, pulmonary arterial hypertension, which is classified by the World Health Organization (WHO) as WHO group 1 pulmonary hypertension [37] is a progressive and fatal disease. The pathobiology of the pulmonary vascular disease in pulmonary arterial hypertension is diverse and not well understood [38], but it is accepted that its pathobiology is programmed in a way to tip the balance in favor of pro-inflammation, pro-coagulation particularly in-situ pro-coagulation, and pro-fibrosis. The regulatory molecules and pathways that are upstream of these pathologic end-points are not well understood or characterized. There is enough evidence now to confirm a major regulatory role of microRNAs in the pathogenesis of pulmonary vascular disease and its clinical prototype, pulmonary arterial hypertension.
Although multiple therapeutic drug families are available to treat pulmonary arterial hypertension [39], their primary effect is via vasodilatation, with only minor other contributory effects such as anti-platelets or anti-inflammatory effects. This may explain the lack of a cure for this disease at this time, and the only modest, but significant, improvement in survival of these patients over the last two decades. As our understanding of microRNAs continues to grow, the field is expected to move forward with new therapeutic pathways that will likely involve targeting microRNAs.
The challenge though, as alluded to above, is that microRNAs do not work in individual sequential pathways. They rather work in concerted efforts among ‘teams’ of microRNAs targeting ‘teams’ of genes. So typically microRNAs are down-regulated or up-regulated in global profiles [40], rather than in individual molecules or individual microRNAs. This biology network requires an eagle’s view of multiple simultaneous and inter-related pathways, and the help of computational network specialists. This should not discourage scientists from going into this very exciting young field, but it has to be tackled with the right mindset. In contrast to our traditional progress in the biomedical fields over the last century, the field of microRNAs will further progress and develop with the use of well-organized and detailed computational and biochemical quantitative models.
Pulmonary Remodeling
Though current treatments for PAH offer a survival benefit, mortality still remains high. Treatments that can effectively control cellular changes and consequent pulmonary remodeling are needed. Hypoxia, vessel injury and inflammation exacerbate the remodeling process [41]. Hyperplastic fibroblasts in the adventitial layer cause adventitial thickening. NADPH increases the reactive oxygen species in adventitia contributing to fibroblast activation. These activated fibroblasts secrete platelet-derived growth factor, endothelin-1 and serotonin causing proliferation, migration and contraction of fibroblasts and smooth muscle cells. A subset of these activated fibroblasts differentiate into myofibroblasts which are regulated by growth factors such as TGF-β [42, 43]. Myofibroblasts migrate into the medial layer increasing its thickness and deposit extracellular proteins such as collagen, fibronectin, elastin and tenascin C which in turn induces smooth muscle cell proliferation and migration within the media [41, 44, 45]. Myofibroblasts and hypertrophic smooth muscle cells considerably increase diameter of the medial layer. Hypertrophy and proliferation of endothelial cells causes intimal thickening leading to increased production of the vasoconstrictor endothelin-1 [46], and elastase which activates matrix metalloproteinases and tenascin-C further increasing smooth muscle cell proliferation. Eventually, increased proliferation of endothelial cells results in formation of plexiform lesions [47], occlusion of the lumen and diminished blood flow to distal pulmonary arteries [48].
Animal Models for PAH
Gomez-Arroyo et al. have extensively reviewed the available mouse models for pulmonary hypertension [49]. The one animal model that most closely resembles the pathobiological hallmarks of pulmonary arterial hypertension in humans is that of the combination of vascular endothelial growth factor receptor antagonist (SU5416) and chronic hypoxia [50]. SU5416 is a combined VEGF receptor 1 (Flt) and 2 (KDR) blocker. This model has been used for preclinical drug testing to determine whether the pulmonary hypertension and pulmonary vascular disease can be reversed once established. Histopathologically, the lung vascular lesions of these rats are comparable to the plexiform lesions in humans [51]. This model has also been tested in various knockout mice which has been reviewed in [52]. Since there is not an ideal mouse model for PAH yet, we cannot be certain that the results seen in pulmonary hypertension animal models can be translated to humans.
MicroRNAs in Pulmonary Hypertension
Profiling studies have identified a characteristic microRNA signature of pulmonary hypertension. Caruso et al. performed microarray analysis in rats exposed to chronic hypoxia and monocrotaline [40]. They found miR-22, miR-30 and let-7f to be downregulated while miR-322 and miR-451 were upregulated. A second study analyzed microRNA expression in pulmonary arteries of PAH patients. Amongst the upregulated microRNAs were miR-138, miR-367, miR-27b, miR-145, miR-302b and miR-450a while miR-204 was downregulated. A few microRNAs have been studied in detail to identify their roles in the pathogenesis of pulmonary hypertension. This is a non-comprehensive list of microRNAs that have been implicated in or associated with the pathogenesis of pulmonary vascular disease and has also been discussed earlier [53]. This list is expected to change rapidly as the field of microRNAs continues to exponentially grow. Here we will discuss each of these microRNAs having divided them into upregulated and downregulated microRNAs in PAH.
Upregulated microRNAs
miR-143 ~ 145: These polycistronic microRNAs are conserved across different species, abundantly expressed in cardiac, vascular and visceral smooth muscle cells [54, 55] to maintain their contractile phenotype. MiR-145 inhibits Kruppel-like factor 5 (KLF5), thus inducing myocardin and smooth muscle cell markers such as α-smooth muscle actin, calpolin and smooth muscle myosin heavy chain [55]. MiR-143 and miR-145 are increased in human PAH and the hypoxia-induced mouse model of PH [56, 57]. PAH patients with BMPR2 mutations have increased expression of miR-145 and in vitro inhibition of BMPR2 induces miR-145. In vascular smooth muscle cells, TGF-β and BMP4 induce myocardin expression and nuclear transport of myocardin-related transcription factors respectively which eventually leads to increased transcription of this polycistron [58]. Inhibition of miR-145 in vivo prevents hypoxia-induced PH in mice [57].
miR-210: MiR-210 is induced by hypoxia in smooth muscle cells and mouse lungs via an HIF-1-dependent pathway. It inhibits E2F3 and increases cellular resistance to apoptosis causing smooth muscle cell hyperplasia [59].
miR-21: MiR-21 is upregulated in lungs of PAH patients and animal models of PH. In pulmonary artery smooth muscle cells, hypoxia induces miR-21 resulting a subsequent decrease in its targets PDCD4 (programmed cell death 4), SPRY2 (sprouty homolog 2) and PPARα. Inhibition of miR-21 inhibits hypoxia-induced proliferation and migration in cultured smooth muscle cells [60] and prevents and reverses hypoxia-induced PAH [61]. Using computational approaches, Parikh et al. identified miR-21 to regulate multiple pathways in PAH [62]. In pulmonary artery endothelial cells, hypoxia and BMPR2 signaling increase miR-21 expression. MiR-21 in turn downregulates BMPR2 expression forming a negative feedback loop. MiR-21 directly targets RhoB expression and Rho-kinase activity decreasing angiogenesis and vasodilatation. Inhibition of miR-21 causes the opposite effects on Rho kinase activity, exaggerating PH in animal models [62]. Decreased miR-21 in mice also activates the PDCD4/caspase-3 axis thus inducing PH. Supplementing miR-21 reduces PDCD4 and protects mice from hypoxia/SU5416-induced PH. There are a few discrepancies in the results reported for miR-21. MiR-21 is downregulated in monocrotaline-induced PH and in endothelial cells of human PAH [40, 63] but a threefold upregulation of miR-21 was observed in hypoxic human pulmonary artery smooth muscle cells, while no change was observed in rats [40, 60]. Caruso et al. demonstrated downregulation of miR-21 in rat lungs of monocrotaline-induced PH but not hypoxia-induced PH, whereas Parikh et al. demonstrated time-dependent induction of miR-21 in monocrotalin-induced PH [40, 62]. MiR-21 prevents PH in the hypoxia/SU5416 model [62, 64] but promotes it in the hypoxia alone model [61]. These contradicting results may possibly stem from the fact that miR-21 has different roles in endothelial cells and smooth muscle cells.
miR-27a: Hypoxia induces miR-27a in pulmonary artery endothelial cells leading to increased cell proliferation and endothelin-1 expression to decrease PPARγ [65]. MiR-27a levels are increased in PPARγ knockout mice, and a PPARγ ligand, rosiglitazone attenuates hypoxia-induced miR-27a. These results suggest a negative feedback loop constituting PPARγ and miR 27a to promote PH
Downregulated microRNAs
miR-17 ~ 92 cluster: The miR-17 ~ 92 cluster includes six mature microRNAs, miR-17, miR-18a, miR-19a, miR19b, miR-20a and miR-92a. This cluster is essential for lung development; loss of this polycistron leads to severe hypoplastic lungs in mice [66]. MiR-17 ~ 92 is downregulated in pulmonary artery smooth muscle cells from PAH patients [67] and associated with reduced expression of α-smooth muscle actin, SM22α and calpolin. Reduced cluster expression is associated with a dedifferentiated smooth muscle cell phenotype which is reversed by overexpression of the cluster. PDLIM5 (PDZ and LIM domain 5) is a direct target of this cluster. In PAH, upregulated PDLIM5 induces the TGFβ3/SMAD3 pathway contributing to PAH [67]. Transient upregulation of miR-17 in the hypoxia-induced model has been reported which was partially reversed by inhibition of miR-17 probably by inducing p21 and subsequent inhibition of proliferation of smooth muscle cells [68]. Hence in the initial stage of blood vessel remodeling, increased miR-17 ~ 92 promotes smooth muscle cell proliferation while in the later stages reduction of this cluster releases its inhibition on its target PDLIM5 to maintain the smooth muscle cell dedifferentiation. In endothelial cells, VEGF induces the miR-17 ~ 92 cluster which is required for endothelial cell proliferation and angiogenesis. A few patients with PAH have BMPR2 mutations and downregulation of BMPR2 is associated with PAH [69]. BMPR2 is a direct target of miR-17-5p and miR-20a in pulmonary artery endothelial cells. The promoter of this polycistron has a conserved STAT3 binding site. IL6 induces STA3 binding to this promoter region, thus increasing expression of miR-17 ~ 92 [70].
miR-124: Kang et al. studied the microRNAs regulating the nuclear factor of activated T cells (NFAT) pathway which has been implicated in PAH and smooth muscle cell proliferation [71]. NFATc1, CAMTA1 and PTBP1 are direct targets of miR-124. Their inhibition results in inhibition of NFAT activity, dephosphorylation, nuclear translocation and inhibition of NFAT-dependent IL2 transcription. MiR-124 is reduced in fibroblasts isolated from PAH patients and animal models of PH [72]. MiR-124 inhibits expression of monocyte chemotactic protein-1 and polypyrimidine tract-binding protein 1 which regulate Notch1, tensin homolog/FOXO3/p21Cip1, p27Kip1 signaling to prevent hyperproliferation and migration of fibroblasts. Histone deacetylase inhibitors restore miR-124 expression and suppress fibroblast proliferation, hinting at a therapeutic role for histone deacetylase inhibitors in PAH.
miR-204: MiR-204 is an intronic microRNA of the gene transient receptor potential melastatin 3 (TRPM3) sharing its promoter with its host gene. MiR-204 is expressed in pulmonary artery smooth muscle cells and downregulated by STAT3 [56, 73]. MiR-204 is reduced in human PAH and hypoxia-induced and monocrotaline-induced PH in rats. Endothelin-1, angiotensin –II, PDGF decrease miR-204; this microRNA reduction in turn activates the Src-STAT3-NFAT pathway by upregulating SHP2 causing smooth muscle cell proliferation and decreased apoptosis. Supplementing miR-204 reverses monocrotaline-induced PH. MiR-204 levels are negatively correlated with severity of PAH in patients and positively correlated with the levels in smooth muscle cells.
miR-424 and 503: An intact apelin signaling pathway is essential for pulmonary vascular homeostasis. In PAH patients, reduced apelin results in increased proliferation and reduced apoptosis of endothelial cells while knockout of apelin exacerbates hypoxia-induced PH [74]. MiR-424/503 are positively regulated by apelin, and they in turn negatively regulate the fibroblast growth factor (FGF) endothelial signaling via their direct effect on FGF2 and FGF receptor-1 [75]. These microRNAs are decreased in PAH. Restoration of miR-424 and miR-503 in rat PAH models improved right ventricular function and PAH pathological molecular and antigen markers by decreasing FGF and its receptor [75].
miR-126: Potus et al. showed decreased capillary density n peripheral skeletal muscles of PAH patients suggesting a defect in peripheral angiogenesis [76]. MiR-126, a microRNA decreased in PAH skeletal muscles and decompensated right ventricle, targets Sprouty-related EVH1 domain-containing protein 1 (SPRED-1) which is a main regulator of the VEGF/ERK pathway. Potus and colleagues also observed an increase in miR-126 and decrease in SPRED-1 in the compensated right ventricle. Supplementing miR-126 restored the angiogenic ability of PAH endothelium to normal levels in quadriceps and right ventricular endothelial cells.
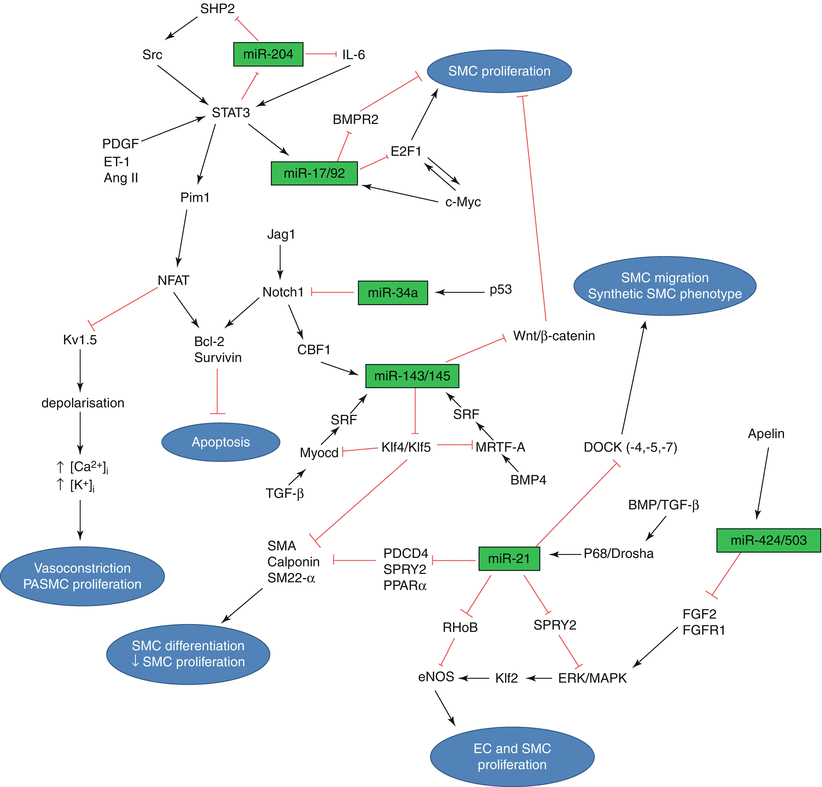
This list of microRNAs is only the tip of the iceberg. A much larger number of yet unidentified microRNAs is expected to be associated or implicated in the pathobiology of pulmonary vascular disease, which includes processes such as pro-inflammation [77], pro-coagulation, pro-proliferation, intimal thickening, medial hypertrophy, and adventitial fibrosis and thickening. These involve a multitude of cells including but not limited to mononuclear cells, platelets, endothelial cell, smooth muscle cells, fibroblasts, and a wide range of cytokines and other transcription factors. These milieus and cells have direct interaction mechanisms with hypoxia [78] and flow-mediated changes. Each of these processes, molecules, milieus, and factors are likely to have their own microRNA profiles changes and networks that will collectively likely involve a much larger number of individual microRNAs. Every single biological, physiologic or pathologic process that explained any step in the pathobiology of pulmonary vascular disease likely has a microRNA regulatory mechanism that is yet to be identified. Grant et al. have summarized the studied microRNAs in the form of a regulatory network [53] (Fig. 7.2). The microRNAs depicted in this figure control pulmonary artery remodeling by targeting genes belonging to different pathways in different cell types. Thus microRNAs form complex regulatory networks by themselves, but since their expression is correlated with their target genes, they form an intertwined network involving multiple pathways. All the molecules in this network participate to maintain homeostasis. Other members of the network may rescue slight perturbations of certain molecules, however sustained excessive perturbations result in disease.
Regulation of microRNAs in Pulmonary Hypertension
The most common mechanism regulating microRNAs in PAH is hypoxia. Hypoxia possibly activates certain transcription factors including hypoxia inducible factors (HIF). Mice heterozygous for HIF1α have reduced pulmonary remodeling [79]. HIF1α induces the expression of miR-210 in endothelial cells [80] and pancreatic cancer cells [81]. Hypoxia induces miR-155 by binding of HIF1α to hypoxia responsive elements in the promoter of miR-155 [82]. HIF1α is a direct target of miR-155 thus creating a negative feedback loop.
The second process which regulates microRNA in PAH is inflammation. During inflammation, numerous cytokines, chemokines and growth factors are released which regulate microRNA expression. For example, in human airway smooth muscle cells, miR-25 is decreased following stimulation with IL-1β, TNF-α and IFN-γ [77]. However, miR-155 and miR-146 are increased by these inflammatory stimuli. MiR-146 is induced by NF-κβ and targets TRAF6 and IRAK1 which activate NF-κβ thus forming a feedback loop [83]. Tissue damage causes NF-κβ nuclear translocation which releases IL-6. IL-6 induces miR-21 transcription in a STAT3-dependent manner [84, 85].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


