and Myron C. Gerson1
(1)
University of Cincinnati, 231 Albert Sabin Way, Cincinnati, OH 45267, USA
Keywords
Sudden cardiac deathImplantable cardiac defibrillatorCardiomyopathy 123I-MIBG 11C-HEDHeart failureSince the 1970s, cardiovascular mortality has decreased significantly as the result of identification and modification of cardiac risk factors, improvement in cardiovascular treatments, and advances in diagnostic imaging. One major advance was the introduction of the implantable cardiac defibrillator (ICD), which substantially increased survival in patients with cardiomyopathy. Unfortunately, however, accurate identification of patients who will develop sudden cardiac death (SCD) remains elusive. Many patients who meet criteria for ICD implantation never require device-administered therapy for ventricular tachyarrhythmias. Conversely, most patients who die suddenly do not meet current guideline criteria for ICD placement before their event. Therefore, a noninvasive test that can identify at-risk patients prospectively would be ideal. Data are mounting showing that radionuclide imaging with iodine-123 metaiodobenzylguanidine (123I-MIBG) for single-photon emission computed tomography (SPECT) or carbon-11 hydroxyephedrine (11C-HED) for positron emission tomography (PET) may provide important prognostic information regarding the development of SCD and might be used to risk stratify patients.
According to current estimates, the incidence of SCD in the United States ranges from 300,000 to 350,000 patients annually [1]. Ventricular tachycardia (VT) and ventricular fibrillation (VF) remain the leading etiology for arrhythmic mortality in patients with heart failure [2]. Initial pharmacologic therapy for SCD was based on the suppression of ventricular arrhythmias identified on Holter monitoring or provocative electrophysiology testing. Although some pharmacologic treatment strategies, such as amiodarone, have been shown to decrease arrhythmogenic deaths [3, 4], other treatments, including encainide, flecainide, moricizine, and sotalol, have been shown to increase mortality [5–7]. Additionally, pharmacologic therapy often is limited by drug toxicity or side effects and has a limited ability to decrease all-cause mortality in post–myocardial infarction patients. The development of ICDs profoundly improved survival for patients with SCD. In patients with ischemic cardiomyopathy in the second Multicenter Automatic Defibrillator Implantation Trial (MADIT II), ICD therapy decreased the relative risk of all-cause mortality by nearly 30 % over 30 months in patients with a left ventricular ejection fraction (LVEF) <30 % [8]. In the Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT), ICD therapy was superior to optimal medical therapy with amiodarone [9]. Based in large part on these and other trials [10–12], current guidelines provide recommendations for an ICD for secondary prevention (after a life-threatening ventricular arrhythmia) or for primary prevention (optimally medically treated patients with New York Heart Association [NYHA] class II or III heart failure and an LVEF <35 % or NYHA class I and an LVEF <30 %) [13]. Although LVEF is an important predictor of SCD [8], it is imperfect, as only 35 % of patients receive appropriate ventricular therapy within 3 years of implantation [14] and most SCDs occur in patients with an ejection fraction >35 % [15].
Some guidelines recommend T-wave alternans, signal-averaged electrocardiography, baroreflex sensitivity (BRS), heart rate variability (HRV), and heart rate turbulence as potentially useful tools for diagnosing ventricular arrhythmias and risk stratifying patients [16]. Additionally, these guidelines recommend use of imaging modalities to investigate underlying structural abnormalities, cardiac function, or ischemia. Unfortunately, these techniques have not been as robust as previously hoped in predicting life-threatening arrhythmias.
Through the use of novel radiotracers, radionuclide imaging may further improve the risk stratification of patients at risk for SCD. This chapter outlines the background of existing imaging methods that have assessed ischemia and scar as markers for SCD. It then reviews the current literature supporting SPECT and PET sympathetic neural imaging for evaluating SCD and ICD placement.
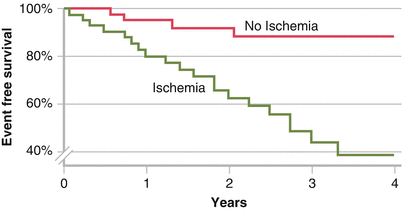
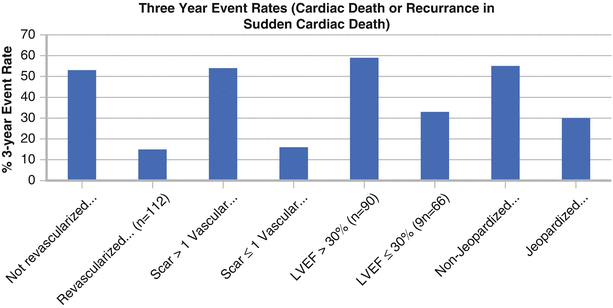
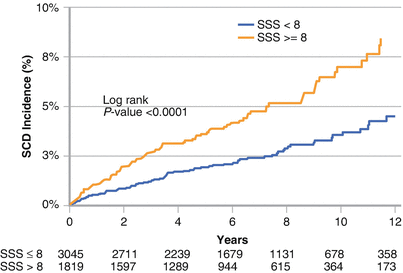
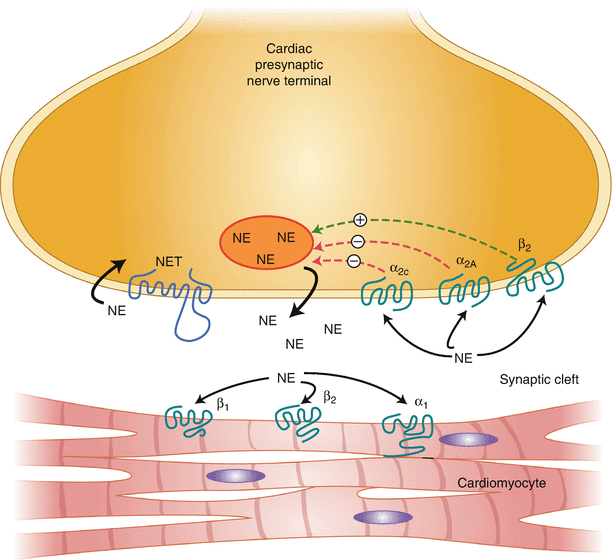
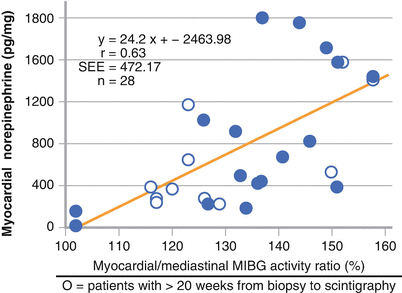
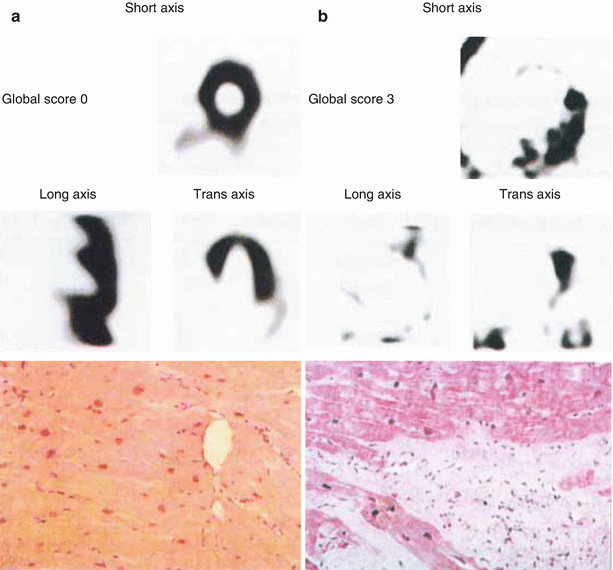
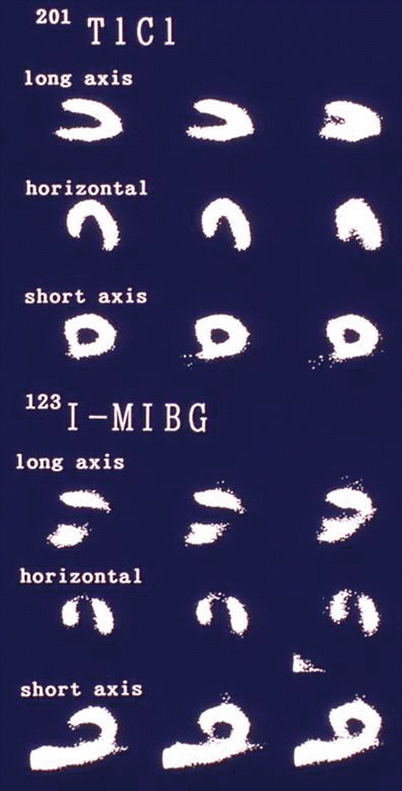
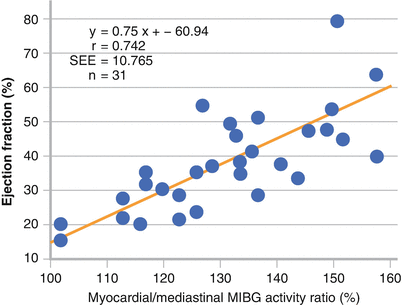
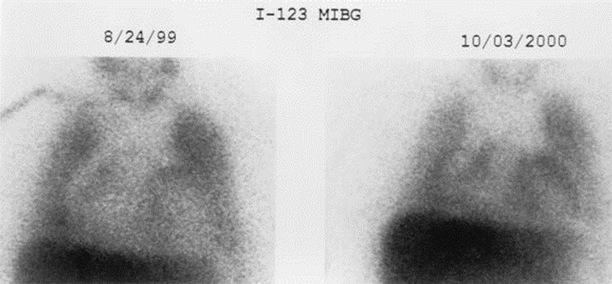
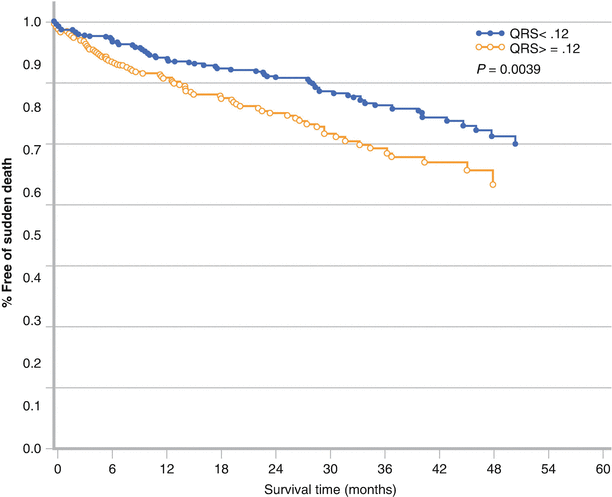
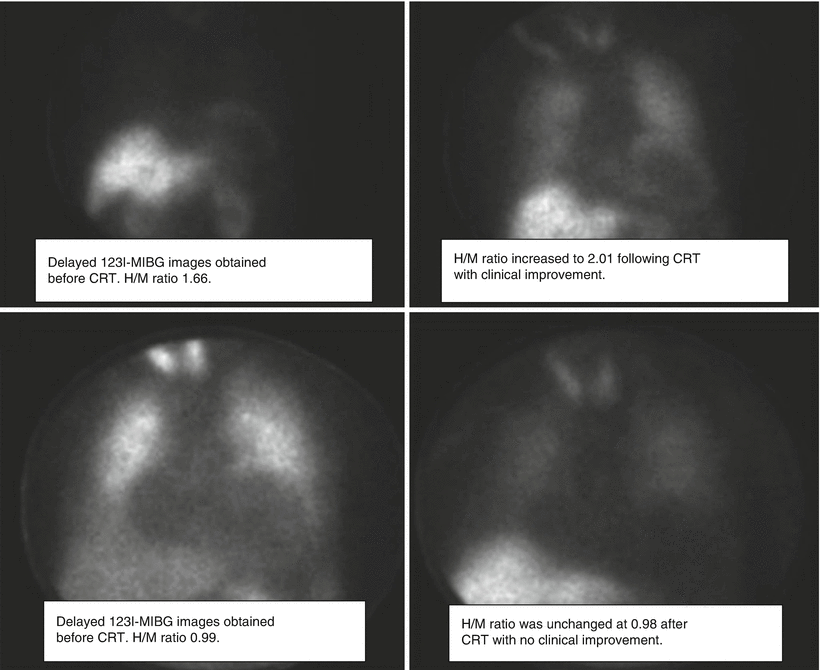
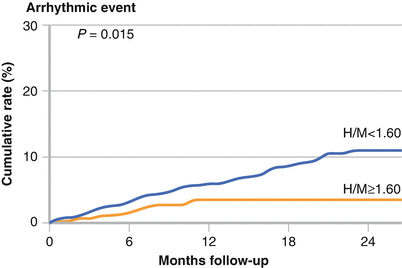
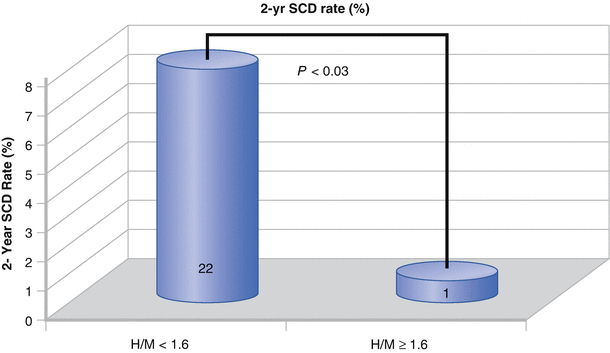
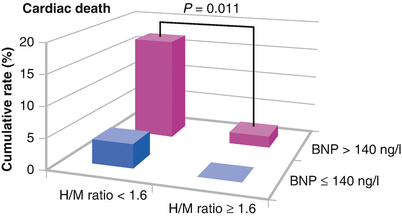
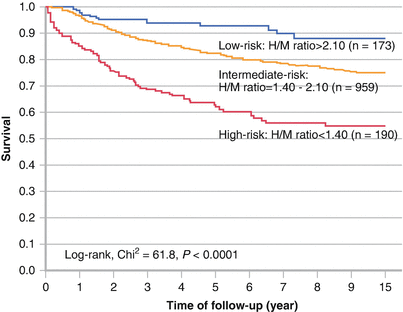
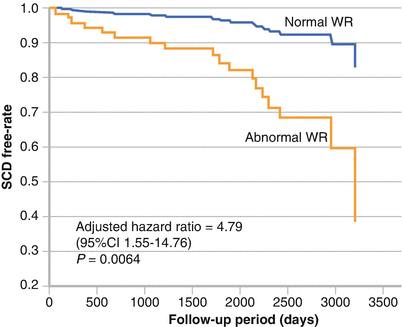
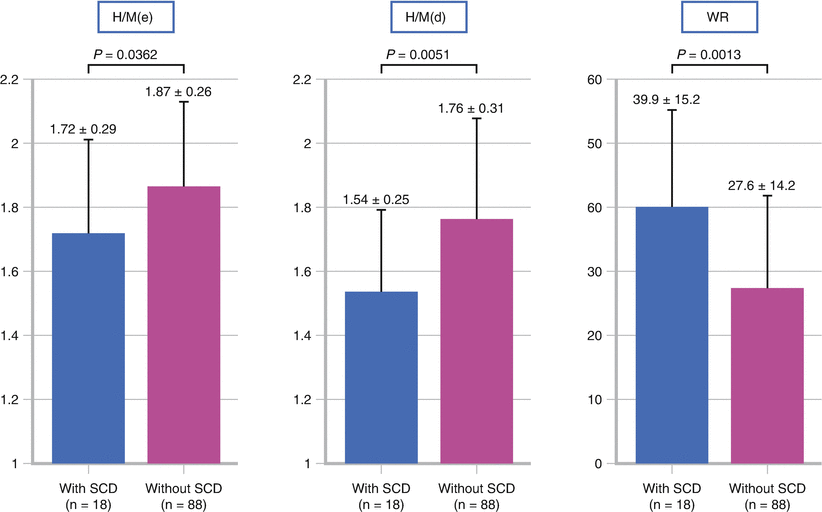
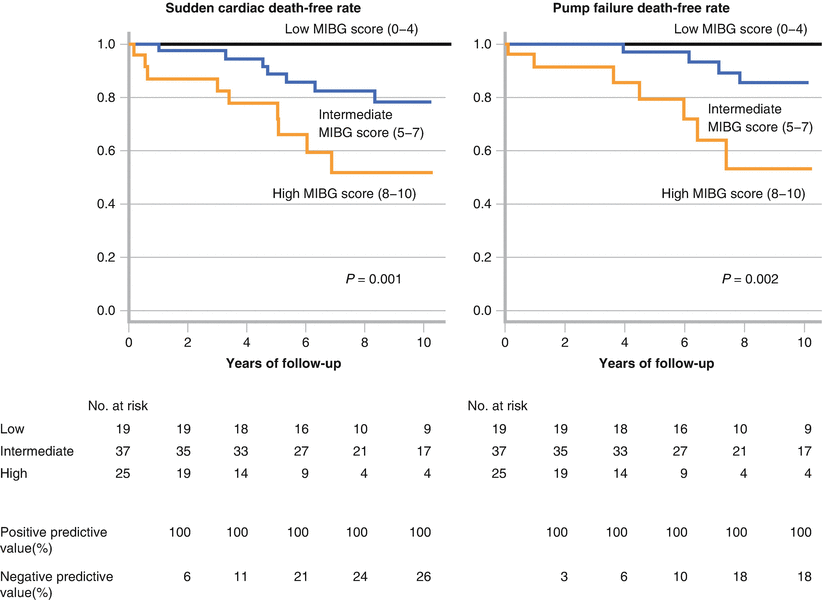
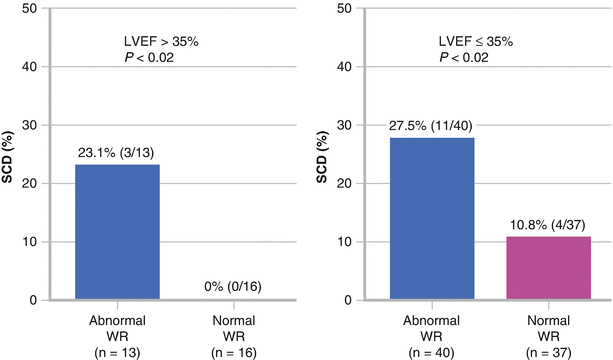
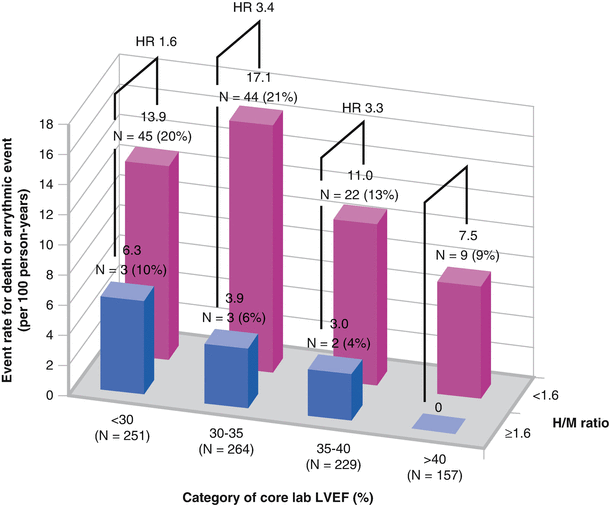
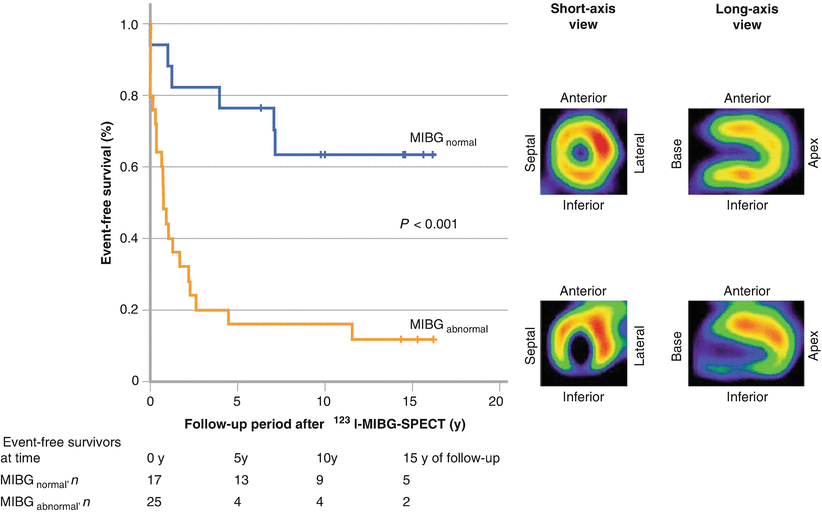

Figure 7-1.
Myocardial ischemia is a well-established risk factor for the development of VT and SCD. In 90 patients who received an ICD, Elhendy et al. [17] showed by Kaplan–Meier analysis that myocardial ischemia detected by stress echocardiography was an independent predictor of SCD or appropriate ICD shock. Yet, myocardial ischemia is only one predictor of SCD. In a meta-analysis of patients with coronary artery disease and left ventricular dysfunction who underwent revascularization [18], those with nonviable myocardium had a significantly higher mortality than those with revascularized viable tissue, suggesting the importance of other factors, including myocardial scar (Adapted from Elhendy et al. [17].).
Figure 7-2.
Myocardial remodeling after an infarction leads to the formation of scar tissue with islands of preserved myocytes. This remodeling allows development of conduction block and re-entrant circuits located within the scar, which act as a nidus for ventricular arrhythmias [19]. Cardiac magnetic resonance imaging (MRI), with its high spatial resolution, has helped detect previously unrecognized scar by using delayed gadolinium enhancement imaging and has been used for risk assessment in patients with or without ischemia [20, 21]. Scar size as determined by MRI is a predictor of VT during electrophysiologic induction testing [22], and the extent of tissue heterogeneity correlates with induction of ventricular arrhythmias [23] and spontaneous ventricular arrhythmias [24]. This figure shows the 3-year rate of cardiac death or recurrent SCD from a SPECT myocardial perfusion imaging study by van der Burg et al. [25]. It characterizes the association of scar burden, presence of jeopardized myocardium, and ejection fraction with cardiovascular events in 153 SCD survivors. On multivariable analysis, the only independent predictors of arrhythmic death were LVEF <30 % and the presence of extensive scar tissue (Adapted from van der Burg et al. [25]).
Figur 7-3.
The combination of a large scar burden and the presence of ischemia has a synergistic effect and increases mortality and SCD [23] compared with either factor alone. In a cohort of more than 6000 patients with coronary artery disease who were evaluated with SPECT myocardial perfusion imaging, Piccini et al. [26] showed that infarct with ischemia, as represented by the summed stress score (SSS), was significantly associated with SCD. Compared with clinical variables, LVEF contributed 48.3 % of the prognostic model content for estimating SCD. By comparison, the combination of ejection fraction and SSS contributed 51.6 % of the prognostic model content for estimating SCD, with the addition of SSS providing a modest improvement in information content. This figure shows Kaplan–Meier curves using a SPECT SSS of 8 in predicting SCD. SSS-predicted SCD was independent of LVEF and clinical variables [27] (Adapted from Piccini et al. [27]).
Figure 7-4.
Although perfusion, scar, and ejection fraction have been beneficial in evaluating patients at risk for SCD, additional discrimination is required because most patients with an ICD never require defibrillation and many with a normal ejection fraction develop SCD. The introduction of 123I-MIBG has facilitated investigation into myocardial sympathetic innervation and function. 123I-MIBG allows direct analysis of cardiac sympathetic function, as it is structurally similar to norepinephrine (NE) and is transported into the cardiac sympathetic neurons by human NE transporter 1 (hNET1), contained on the presynaptic nerve terminus [28]. 123I-MIBG requires an intact cardiac sympathetic nervous system for normal uptake, is stored in the presynaptic vesicles, and is released by stimulation from NE [29]. Experimental manipulation of cardiac sympathetic function alters MIBG uptake and distribution [30–33]. In both ischemic and dilated cardiomyopathy, increased sympathetic nerve activity results in a decrease in the number and function of myocardial β-adrenergic receptors [34, 35]. Chronically increased catecholamine levels trigger a cascade of events leading to altered β-adrenergic receptor regulation, impaired NE reuptake and calcium regulation, depletion of presynaptic NE stores, and cardiac fibrosis and remodeling. This figure illustrates the synaptic cleft, showing the uptake and regulation of NE. 123I- MIBG follows the same uptake and regulation as those of NE, but is not metabolized by monoamine oxidase or catechol-O-methyltransferase.
Figure 7-5.
Planar image acquisition after 123I-MIBG injection enables evaluation of sympathetic activation. Early and delayed measurement of the heart-to-mediastinum (H/M) ratio assesses the initial uptake and washout of the tracer. In 28 patients with NYHA I to IV dilated cardiomyopathy, the H/M ratio correlated with myocardial NE concentration on cardiac biopsies. SEE standard error of the estimate (Adapted from Schofer et al. [36]).
Figure 7-6.
Fibrotic myocardial tissue has reduced neuronal NE content and decreased 123I-MIBG uptake. Murata et al. [37] showed that 123I-MIBG uptake in 24 patients with dilated cardiomyopathy, as measured by a 0 to 3 grading system for normal to no uptake, respectively, correlated with the level of histologic abnormalities, including myocyte hypertrophy, myocardial fibrotic change, myocyte degeneration, mononuclear cell infiltration, and myocyte disarray [37]. This figure shows two patients, one with normal uptake and a global score of 0 (a) and one with severely decreased uptake and a global score of 3 (b) (From Murata et al. [37]; with permission).
Figure 7-7.
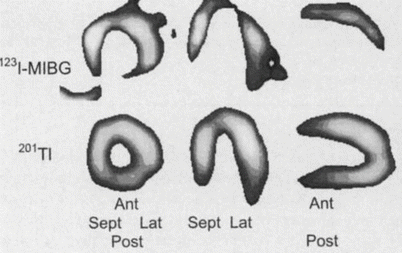
A substrate for ventricular arrhythmias in patients with coronary artery disease is the presence of myocardium that is denervated but remains viable after an acute coronary event. The extent of decreased myocardial 123I-MIBG uptake has been shown to extend beyond the infarcted territory and to be present after an ischemic insult without persistent perfusion abnormalities [38]. This innervation–perfusion mismatch likely represents the at-risk myocardium and has been characterized as a marker for myocardial ischemic memory [39]. These areas of decreased 123I-MIBG uptake represent denervated myocardium, which is at an increased risk for augmented early afterdepolarizations in response to catecholamines [40] and increased arrhythmogenicity [41]. As a marker of myocardial fibrosis, reduced 123I-MIBG H/M ratio has been correlated with reduced LVEF [36]. With its relationship to myocardial fibrosis, LVEF, peri-infarction ischemia, and electrically abnormal myocardium, the extent and distribution of reduced 123I-MIBG in the myocardium has consistently predicted SCD. The figure displays thallium myocardial perfusion images and 123I-MIBG images obtained after resolution of a 6-h episode of ischemic chest pain. The perfusion images are normal, but the 123I-MIBG images show a defect predominantly in the apical, anterior, and inferior walls. On left heart catheterization, there were 75 % left anterior descending and right coronary artery lesions [39]. 201 TlCl thallium-201 chloride (From Tomoda et al. [39]; with permission).
Figure 7-9.
During recent decades, the mortality from heart failure has decreased because of earlier and better therapies, including pharmacologic treatments, revascularization, and resynchronization. Intuitively, as clinical status improves with treatment, MIBG uptake and washout also should improve. In 2002, Gerson et al. [42] studied 22 patients with idiopathic cardiomyopathy by using 123I-MIBG scintigraphy before and after carvedilol treatment. Ten patients had severe myocardial sympathetic impairment with an H/M ratio <1.4. In this group, LVEF increased from 25.4 to 37.3 % with a concomitant increase in H/M ratio from 1.26 to 1.39 [42]. Other investigators reported similar responses with β-blockers [43, 44]. Improvement in 123I-MIBG H/M ratio also has been documented in response to antiarrhythmic therapy with amiodarone [45–47]. Whether the presence or absence of a favorable response of cardiac neuronal measurements to guideline-based heart failure therapies can stratify risk for sudden cardiac death needs further documentation. Shown here are planar 123I-MIBG images from a patient with heart failure before and after treatment with carvedilol. There is a visually apparent increase in myocardial 123I-MIBG uptake 10 months after the initiation of treatment. H heart, M mediastinum (From Gerson et al. [42]).
Figure 7-10.
Patients with systolic heart failure, wide QRS complexes, and a left bundle branch block pattern are at an increased risk for SCD and overall mortality compared with patients with heart failure and a narrow QRS complex [48]. Cardiac resynchronization therapy (CRT) has been shown to lead to a reduction in SCD and a 36 % relative risk reduction in overall mortality, compared with medical therapy alone [49] (Adapted from Iuliano et al. [48]).
Figure 7-11.
Delayed 123I-MIBG images obtained before and after CRT. In the patient in the upper images, who had a favorable clinical response to resynchronization therapy, the H/M ratio before CRT was 1.66 (left) and increased to 2.01 after resynchronization (right). The lower images are from a patient with no clinical improvement after CRT, with an H/M ratio of 0.99 before (left) and 0.98 after therapy (right). Nishioka et al. [50] showed that the 123I-MIBG H/M ratio and washout rate are associated with response to resynchronization therapy and that the H/M ratio may be useful in predicting which patients will respond to CRT (From Nishioka et al. [50]).
Figure 7-12.
The AdreView Myocardial Imaging for Risk Evaluation in Heart Failure (ADMIRE-HF) study investigated the potential role of 123I-MIBG H/M ratio in predicting life-threatening ventricular arrhythmias [51]. This large prospective trial used a 123I-MIBG H/M ratio of less 1.6 for predicting cardiovascular events in patients with symptomatic heart failure. The study enrolled 961 patients (NYHA class II–III) with severe systolic heart failure (LVEF ≤35 %). More than 90 % of the patients were receiving β-blockers, angiotensin-converting enzyme inhibitors, or angiotensin receptor blockers. Those with a ventricular pacemaker, history of defibrillation, ICD, acute myocardial infarction, or serum creatinine >3.0 mg/dL were excluded. LVEF was measured primarily by echocardiography at the enrolling site. The patients were divided into two prespecified groups (H/M ratio >1.6 and H/M ratio <1.6). Eighty-six participants had major arrhythmic events: 63 nonfatal events, including sustained VT, aborted cardiac arrest, and ICD firing, and 23 SCDs. The Kaplan–Meier curves show a significant increase in the rate of major arrhythmic events for patients with an H/M ratio <1.6 (Adapted from Jacobson et al. [51]).
Figure 7-13.
Sudden cardiac death rate during 2-year follow-up. SCD occurred in 22 subjects with an H/M ratio <1.6 and in only 1 subject with an H/M ratio ≥ 1.6 [51] (Adapted from the ADMIRE-HF study, courtesy of Arnold Jacobson MD, PhD).
Figure 7-14.
In ADMIRE-HF, the 123I-MIBG H/M ratio provided additional risk stratification for cardiac death when compared with a brain natriuretic peptide (BNP) level >140 ng/L or LVEF <30 % and was an independent predictor of cardiac death. The Kaplan–Meier survival curves in this figure depict the significantly increased rate of cardiac death for patients with BNP >140 ng/L and an H/M ratio <1.6. In those with BNP <140 ng/L the addition of H/M ratio did not predict an increased event rate [51] (Adapted from Jacobson et al. [51]).
Figure 7-15.
Sudden cardiac death and progressive heart failure are responsible for most deaths in patients with severe heart failure and left ventricular dysfunction. Nakata et al. [52] pooled data from six prospective cohort trials involving 1322 patients to investigate all-cause mortality in patients with heart failure. Mean follow-up was 5 years, and the mean LVEF was 37 %. In this cohort, 326 patients had a lethal event, and the mortality rate was 5.6 %, 11.3 %, and 19.7 % at 1-, 2-, and 5-year follow-up, respectively. Based on a multivariate Cox model, age, NYHA class, late H/M ratio, and LVEF were independent predictors of survival. An analysis separating the cohort into three groups—low risk, H/M >2.1; intermediate risk, H/M 1.4–2.1; and high risk, H/M <1.4—showed a significant decrease in survival with worsening MIBG late H/M ratio. Thresholds of 1.68 for H/M ratio and 43 % for the washout rate were determined by receiver operator analysis to predict an increased risk for all-cause mortality (P < 0.0001 for both) (Adapted from Nakata et al. [52]).
Figure 7-16.
In addition to H/M ratio, the washout rate (WR) is an important predictor of SCD. Myocardial washout of 123I-MIBG is defined by the following equation: early 123I-MIBG counts per pixel minus late 123I-MIBG counts per pixel divided by early 123I-MIBG counts per pixel. Tamaki et al. [53] followed up 106 consecutive heart failure patients with an LVEF <40. SCD was defined as witnessed cardiac arrest and death within 1 h of the acute onset of symptoms. An abnormal MIBG washout rate, defined as <27 %, was a significant predictor of SCD after adjustment for age, gender, and LVEF. Signal-averaged ECG, HRV, and QT dispersion were not independent predictors of SCD in this study. Furthermore, 123I-MIBG washout rate may be used to further risk stratify patients with stable cardiomyopathy who are on medical therapy [54]. The 123I-MIBG washout rate in patients with chronic heart failure was shown to be an independent predictor of SCD and provided additional risk stratification when compared with the Seattle Heart Failure Model alone [55] (Adapted from Tamaki et al. [53]).
Figure 7-18.
It has been suggested that both the 123I-MIBG delayed H/M ratio and the washout rate provide additive value in predicting SCD. Kawai et al [56] classified patients into low-, medium-, and high-risk groups based on the sum of the deviation of both H/M ratios and washout rates from the mean values in a control population. They studied 81 patients with NYHA class I, II, or III heart failure and an LVEF <35 %. No patients had an ICD or biventricular pacemaker at study entry. Exclusion criteria included renal dysfunction, insulin-dependent diabetes mellitus, and autonomic neuropathy. During a mean follow-up of 6.9 ± 3.1 years, 16 SCDs occurred, but none of the 19 patients in the low-risk group died (P = 0.001). The authors concluded that an MIBG score including H/M ratio and washout rate may identify heart failure patients at very low risk for SCD (Adapted from Kawai et al. [56]).
Figure 7-19.
Patients with cardiomyopathy and an LVEF >35 % require special attention because they do not meet the criteria for primary prevention ICD implantation but represent most patients who have SCD. 123I-MIBG data are limited in this patient population. The figure shows patients with an LVEF >35 % (left) and those with an LVEF ≤35 % (right) grouped by a normal (<27 %) or an abnormal (>27 %) washout rate (WR). An abnormal washout rate was shown to be significantly associated with SCD by the log-rank test, regardless of LVEF (P values are shown above) (Adapted by Tamaki et al. [53]).
Figure 7-20.
A subgroup analysis of the ADMIRE-HF trial showed a significant SCD association for patients with an H/M ratio <1.6, regardless of baseline LVEF [57]. Although LVEF ≤35 % was an inclusion criterion for ADMIRE-HF, significant interobserver variability exists. On core laboratory review, 386 of 901 patients whose LVEFs were available for evaluation were found to have an LVEF >35 % (overall mean, 34 % ± 7 %). The ADMIRE-HF patients were then grouped according to LVEF and H/M ratio <1.6 (high risk) or ≥1.6 (low risk). Regardless of ejection fraction, the risk of death or arrhythmic events was significantly increased (Adapted by Shah et al. [57]).
Figure 7-21.
Although ischemic and nonischemic cardiomyopathy are common causes of life-threatening ventricular arrhythmias, channelopathies such as arrhythmogenic right ventricular cardiomyopathy (ARVC), Brugada syndrome, and congenital long QT syndrome require specific attention. ARVC is a predominantly autosomal dominant inherited cardiomyopathy afflicting 1 in 5000 people [58]. Pathophysiologic characteristics, including myocardial fatty infiltration and fibrosis, result in nonhomogeneous refractory periods and provide a substrate for ventricular arrhythmias and SCD. This arrhythmogenic potential is exacerbated further by catecholamines, which increase the refractory dispersion. In some series, ARVC accounted for approximately 22 % of SCDs in athletes [59]. Current guidelines recommend ICD implantation for secondary prevention in ARVC patients with VT or VF and for primary prevention in patients with extensive disease, left ventricular involvement, a family history of SCD, or undiagnosed syncope [16]. Screening is imperfect, and additional techniques are needed for risk stratification in this high-risk population. The figure shows representative 123I-MIBG images [60] from a control subject (upper right) and a patient with ARVC (lower right) with decreased inferoseptal activity. The survival curves (left) show significantly decreased event-free survival for ARVC patients with an abnormal scan (Adapted from Paul et al. [60]).
Figure 7-22.
In patients with channelopathies, it has been postulated that heterogeneous cardiac sympathetic innervation contributes to life-threatening ventricular arrhythmias. In a canine model, Dae et al [33] showed that stellate nerve ablation can produce regional variation in sympathetic innervation that is independent of regional myocardial perfusion. The left stellate ganglion innervates the right ventricle and the left ventricular inferior and inferoseptal walls, whereas the right stellate ganglion innervates the anterior and lateral walls. The figure shows MIBG and thallium-201 dual-isotope SPECT images after stellectomy. Equal innervation and perfusion are displayed in red, decreased MIBG relative to perfusion in yellow to green, and increased innervation relative to perfusion in blue. The myocardial slices show denervation of the inferior and inferoseptal left ventricle after left stellectomy and anterior denervation of the left ventricle after right stellectomy (From Dae et al. [33]; with permission).
Table 7-1.
Neuronal imaging reported findings in ion channel disorders linked to sudden cardiac death.
Clinical disorder | Onset | Global norepinephrine reuptake | β-Receptors | Regional reuptake |
|---|---|---|---|---|
Stress | ↓ by MIBG, trend only by PET | ↓ by PET | ↓ by MIBG inferoseptal walls, trend only by PET | |
Rest | ↑ by PET but not by MIBG | No change | ↓ by MIBG inferoseptal walls, trend only by PET | |
Stress | ↓ or no change | No data | ↓ anteroseptal ± lateral in most but not all reports | |
Stress | ↓ | ↓ | ↓ inferior or septal by MIBG, uniform by PET |