Vasoconstriction
Vasodilation
Angiotensin II
Natriuretic peptides
Arginine-vasopressin
Kinin-kallikrein system
Catecholamines
Medullipine system
Endothelin
Adrenomedullin
Prostaglandins
Estrogens
Insulin
Nitric Oxide
10.2.1 Renin-Angiotensin System
The systemic renin-angiotensin system (RAS) is central to the regulation of fluid balance. Hyponatremia, hypovolemia, hypotension, and adrenergic stimuli activate the production and release of renin, which enzymatically induce the production of angiotensin I (inactive) that is converted to the vasoactive angiotensin II (Ang II) by a nonspecific converting enzyme (ACE) (Fig. 10.1) [16]. Ang II brings about a constriction of the arteriolar vascular smooth musculature and a release of aldosterone from the adrenal cortex with a resultant increase in tubular sodium and water reabsorption. Since hypervolemia, hypernatremia, and increases in blood pressure and Ang II concentrations, under physiological conditions, supplant renin activation through a negative feedback mechanism, this system may represent a regulatory circuit serving fluid and blood pressure homeostasis. Ang II is able to potentiate SNS activity at different levels [8]. Experimental studies have shown that intracerebral infusion of Ang II was able to induce hypertension, systemic vasoconstriction, and baroreflex reset towards higher blood pressure levels [17]. It has been also demonstrated that vascular smooth muscle nicotinamide dinucleotide phosphate (NADPH) oxidase is activated by Ang II and this increases vascular reactive oxygen species (ROS) levels [18]. More recently, it has been shown that a similar phenomenon occurs within the central nervous system where Ang II upregulates NADPH oxidase activating type 1 (AT1) receptors in neuroanatomical areas implicated in central sympathetic regulation such as the rostroventrolateral medulla (RVLM) in the bulbar area, the circumventricular organs, and the paraventricular nuclei [19–21]. At the peripheral level, Ang II facilitates neural transmission within sympathetic ganglia; favors norepinephrine release by sympathetic nerve terminals, acting on presynaptic receptors; and enhances α-mediated vasoconstriction in arterioles [8, 17, 22–25] (Fig. 10.2). Ang II can stimulate the synthesis of the constitutive enzyme nitric oxide synthase (eNOS) and enhance the production of NO [26, 27]. In the renal circulation, this Ang II-NO interaction may protect the preglomerular vessels from the constrictor effect of Ang II [28]. In patients with coronary artery disease, ACE inhibitors attenuate sympathetic coronary vasoconstriction, not only when the drugs are systematically administered [29], but also when small doses of these compounds are infused at the level of the coronary circulation [30]. This finding suggests that not only systemic but also the local renin-angiotensin system is important for preserving cardiovascular homeostasis via an interaction with endothelial factors.
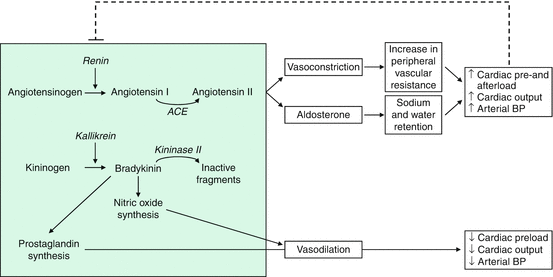
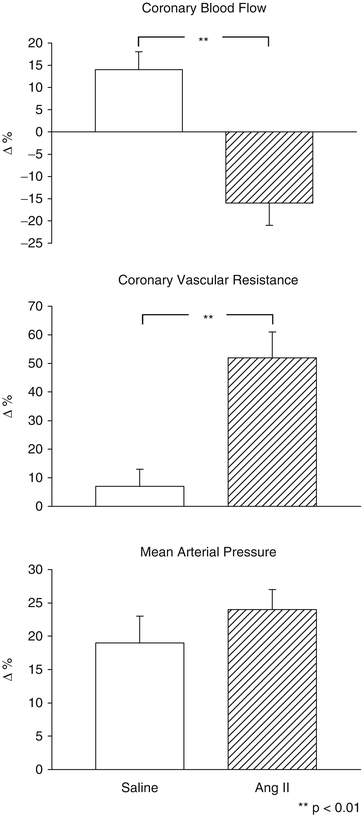
Fig. 10.1
Effects of the renin-angiotensin-aldosterone system and the renal kinin system and their interactions. ACE angiotensin converting enzyme, BP blood pressure, dashed line indicates the inhibition of renin secretion through negative feedback
Fig. 10.2
Changes in coronary hemodynamics and systemic blood pressure induced by diving during the intracoronary infusion of saline and angiotensin II (Data from Saino et al. [25])
10.2.2 Endothelin
Endothelin-1 (ET-1) is a vasoconstrictor peptide produced by endothelial cells with an important role in regulation of vascular tone [31]. The role of the ET system in cardiovascular homeostasis is not limited to its direct vascular effects but also involves the neural regulation of vasomotor tone [32]. ET-1 can stimulate central and peripheral SNS activity through ETA receptors [33, 34]. The mechanism of ET-mediated vasoconstriction involves binding to specific receptors on vascular smooth muscle and direct activation of voltage-operated calcium channels in vascular smooth muscle membrane. Two distinct ET receptors have been identified. The ETA receptor is expressed in vascular smooth muscle cells, whereas the ETB receptor has been localized to the endothelial and smooth muscle cells. The ETB receptor may have a dual vasoconstrictive and vasodilatory effect. While in vivo selective ETA receptor antagonists causes forearm vasodilatation in resistance vessels due mostly to increased NO generation, stimulation of ETA receptors triggers acute vasoconstriction of large conduit arteries [33]. The ETB receptor antagonism causes local vasoconstriction, indicating that these receptors in blood vessels respond to ET-1 majorly causing vasodilation [34]. While intracerebral administration of ET-1 can increase BP and SNS activity mainly through ETA receptors both in hypertensives and normotensives, the administration of an ETA receptor antagonist determines the opposite effect only in hypertensives, suggesting a specific sympathoexcitatory role for the endogenous ET system in this condition [35, 36]. The increased ET-1 vascular tone observed in essential hypertension seems to be a consequence of reduced NO availability, due to the complex interactions between endothelium-derived substances [37]. At the peripheral autonomic nervous system level, ET-1 can act in carotid bodies and in cervical superior and nodose ganglia, influencing baroreflex and chemoreflex regulation. ET-1 is also released by postganglionic sympathetic neurons, modulating catecholamine release and vascular tone, and stimulates catecholamine release from adrenal gland [38].
10.2.3 Arginine-Vasopressin
The peptide hormone arginine-vasopressin or antidiuretic hormone (ADH) is produced in the hypothalamus and stored in the neurohypophysis. The hormone release is induced by hyperosmolarity and volume decrease and causes a rise in iso-osmotic reabsorption of primary urine in the distal tubule and collecting duct of the kidney. It has also been proven in isolated vascular preparations that vasopressin has a strong vasoconstrictor effect [39].
10.2.4 Natriuretic Peptides
The atrial-, brain-, and C-type natriuretic peptides (ANP, BNP, CNP) are a family, structurally similar, with natriuretic and diuretic properties.
ANP, formed and stored in the atrium and released into the plasma through atrial stretching, is a circulatory peptide hormone which may be involved in the regulation of sodium homeostasis and blood pressure [40]. Under physiological conditions, it is possible to detect relatively constant ANP plasma levels which, considering the high clearance rate of this hormone, suggests a continuous and relatively high production rate. In the 1990s, several papers have shown a complex interaction between this substance and the major reflex mechanisms responsible for cardiovascular homeostasis. Animal studies have shown that the reduction in blood pressure that follows the administration of natriuretic peptides is not accompanied by the expected tachycardia [41], which suggests that these substances may produce an activation of the arterial and/or cardiopulmonary baroreflexes. Direct demonstration of ANP-induced potentiation of the baroreceptor-heart rate reflex has been provided in conscious normotensive rats [42]. Thoren et al. [43] have also shown the link with cardiopulmonary reflex activation providing that these peptides were able to stimulate vagal inhibitory fibers in the cardiopulmonary region.
BNP is found predominantly in the ventricles and atria of the heart [44]. Infusion of BNP, as well as ANP, is able to induce natriuresis and diuresis. Increased plasma levels of this peptide hormone, along with higher intravascular volume and increased ventricular pressure, have been observed in several pathophysiological conditions like congestive heart failure, pulmonary hypertension, arterial hypertension, cerebrovascular diseases, liver dysfunction, and chronic kidney disease [45–49]. A role of cardiac natriuretic peptides has been recently shown also in inflammation and rheumatic diseases [50, 51].
In contrast to ANP and BNP, whose primary target organ is the kidney, C-type natriuretic peptide appears primarily to be a paracrine hormone involved in the regulation of vascular tone. CNP is formed from and secreted by vascular endothelium and has been detected in various areas of the brain. It has been proposed to mediate vascular relaxation by causing endothelium-dependent hyperpolarization. To study this in porcine coronary arteries, the endothelium-dependent relaxation and hyperpolarization of CNP and bradykinin were compared. In contrast to bradykinin, CNP induced endothelium-independent and weaker relaxation and hyperpolarization of coronary artery vascular smooth muscle, suggesting that it is an unlikely mediator of endothelium-dependent hyperpolarization of porcine coronary arteries [52].
10.2.5 The Kinin-Kallikrein System and Prostaglandins
Kinins are generated from precursor known as kininogens by enzymes such as tissue and plasma kallikrein (Fig. 10.1). Some of the effects of kinins are mediated via autocoids such as eicosanoids, NO, endothelium-derived hyperpolarizing factor (EDHF), and/or tissue plasminogen activator (tPA). Kinins, acting via NO, help protect against cardiac ischemia contributing to the vascular protective effect of ACE inhibitors during neointima formation and help reduce infarct size following preconditioning or treatment with ACE inhibitors. This mechanism is also useful in heart failure secondary to ischemic heart diseases. In the kidney, kinins are essential for proper regulation of papillary blood flow and water and sodium excretion. Kinins are involved in the acute antihypertensive effects of ACE inhibitors but not their chronic effects (save for mineralocorticoid-salt-induced hypertension). Kinins appear to play a role in the pathogenesis of inflammatory diseases such as arthritis and skin inflammation; they act as mediators of inflammation by promoting maturations of dendritic cells, which activate the body’s adaptive immune system and stimulate mechanisms that promote inflammation. Several prostaglandin derivates (prostacyclin, PGE2), whose synthesis can be stimulated by kinins, are likewise classified as vasoactive, dilatory tissue hormones [53–55].
10.2.6 Medullipin System
Medullipin is a substance formed and released in the renal medulla and converted in the liver to the vasodilatory, blood pressure-decreasing, circulating hormone medullipin II. It lowers intrarenal vascular resistance and increases renal plasma flow, the glomerular filtration rate, diuresis, and natriuresis [56].
10.2.7 Adrenomedullin
Endogenous adrenomedullin is an autocrine or paracrine factor in cardiovascular and renal regulation. This newly identified peptide is widely distributed in human tissue. Adrenomedullin is a potent vasodilator in vivo in both human and animal arteries and, at least in vitro, inhibits cell proliferation and migration as well as ET-1 and Ang II production and oxidative stress. Adrenomedullin exerts cardiovascular protective actions, such as decreasing blood pressure, reducing intima hyperplasia, and is involved in the regulation of in situ vascular proliferations and vascular remodeling, as well as vascular tone. In the kidney, adrenomedullin elicits a dose-dependent increase in renal blood flow, glomerular filtration rates, and urinary sodium excretion. Thus, adrenomedullin plays a role in the physiological regulation of renal hemodynamic and in pathological condition such as salt-sensitive hypertension [57, 58].
10.2.8 Estrogens
Both endothelial and vascular smooth muscle cells possess estrogen receptors. Estrogens regulate the transcription of numerous genes, and its cellular actions are mediated through the translation of specific mNA transcripts and synthesis of proteins. It stimulates the native synthesis of NO in the blood vessels, heart, and skeletal muscles [59, 60]. Estrogens enhance the binding activity of the transcription factor Spl, whose function is essential for eNOS transcription. Even modest increases in eNOS expression may display protective effects against cardiovascular disease [61]. Other potential protective mechanisms of estrogen are represented by the suppression of a prostaglandin H synthase-dependent vasoconstriction [62] and the inhibition of cyclooxygenase-dependent production of oxidative stress [63]. In perimenopausal women, estrogen supplementation reduces arterial blood pressure and enhances basal NO release in forearm resistance arteries [64].
10.2.9 Insulin
Insulin is synthetized and secreted by the pancreas. Although the liver is the main source of circulating IGF-I levels, it is formed in endothelial and vascular smooth muscle cells. The net effects of insulin on the vasculature are determined by different cellular signaling pathways that are activated by stimulation of the insulin receptor (IR) (Fig. 10.3) [65, 66]. Insulin metabolic signaling results in vasodilation via increased NO production and increases in bioavailable NO. However in conditions of insulin resistance, it promotes vasoconstriction and vascular proliferation. Binding of insulin to IR triggers its phosphorylation and activation via an intrinsic kinase activity, leading to tyrosine phosphorylation of the insulin receptor substrate (IRS) proteins. The activation of the PI3K-Akt pathway induces phosphorylation of eNOS and production of NO, modulated in a Ca2+/calmodulin-sensitive manner, with consequent decrease in vascular tone and vascular smooth muscle cell (VSMC) proliferation and reduction in adhesion of inflammatory cells and platelet aggregation to endothelium [67]. Insulin also promotes, with different eNOS phosphorylation, the production of reactive oxygen species (ROS) and modulates the production of prostaglandins and endothelium-derived hyperpolarizing factors. In addition to vasodilation, insulin can promote vasoconstriction. Under some circumstances, insulin activates the mitogen-activated protein kinase (MAPK) cascade that coordinates insulin vasoconstriction and growth-promoting effects [66]. These effects are mediated in part by the increased production of ET-1 and the activation of signaling through the vascular tissue RAAS [65, 66]. ET-1, via ETA receptors, causes vasoconstriction, increases oxidative stress, and promotes cell growth and mitogenesis in VSMCs [33]. Similar deleterious effects are seen with the activation of vascular tissue RAAS [66]. Insulin plays a modulatory role on skeletal muscle vasculature by increasing blood flow and regulating its own delivery during increased metabolic demands [68]. The mechanism is dependent on an intact endothelium and involves activation of the α-adrenergic receptors to restrict the vasodilation to actively contracting areas of the muscle. Once the terminal arterioles are maximally dilated, adequate skeletal muscle capillary recruitment is needed. An impaired capillary recruitment is present in clinical conditions characterized by insulin resistance such as obesity and type 2 diabetes mellitus [68, 69].
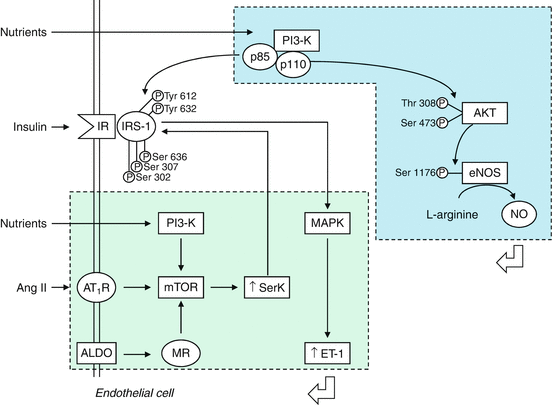
Fig. 10.3
Insulin effects on endothelial cells. Stimulation of IR results in activation of the PI3K-Akt pathway, eNOS phosphorylation, and vasodilation. Insulin resistance induced by RAAS activation and excess nutrients causes increased serine phosphorylation of insulin receptor substrate and metabolic signaling with uninhibited activation of mitogenic and growth pathways. ALDO aldosterone, Ang II angiotensin II, AT1R angiotensin II type 1 receptor, eNOS endothelial NO synthase, ET-1 endothelin 1, IR insulin receptor, mTOR mammalian target of rapamycin, MR mineralcorticoid receptor, MAPK mitogen-activated protein kinase, NO nitric oxide, PI3K phosphatidylinositol 3 kinase, p phosporylation, Akt protein kinase B, Ser serine, SerK serine kinase, thr threonine, Tyr tyrosine (With permission from Muniyappa and Yavuz [66])
10.3 Endothelial Factors
NO is produced from the amino acid L-arginine by the enzyme NO synthase (NOS) which is present in three isoforms [70]: neuronal NOS (nNOS) is expressed in neurons in central and peripheral nervous system and also in macrophages and endothelial cells where it plays a role in regulation of basal vascular tone [71]; inducible NOS (iNOS) is expressed in different cellular types and its activity is induced by inflammatory stimuli [70]; endothelial NOS (eNOS) is a constitutive enzyme isoform found in endothelial and neural cells. NO release from endothelium is determined by receptor-mediated mechanisms (acetylcholine, bradykinin, serotonin, substance P, adenosine diphosphate) and by mechanical stimuli (shear stress) [72]. eNOS expression is negatively influenced by hypoxia, tumor necrosis factor-α, inflammatory cytokines, and by false substrates such as N-monomethyl-L-arginine (L-NMMA), which are commonly used to test the degree of endothelium-dependent vasodilation [70]. Asymmetric dimethylarginine (ADMA), a naturally occurring amino acid, is an endogenous inhibitor of eNOS, which can cause endothelial dysfunction and is associated with increased cardiovascular risk [72]. Several evidences have shown that NO acts as a sympathoinhibitory substance within the central nervous system [9]. It is well established that acute or chronic administration of exogenous NOS inhibitors are able to induce vasoconstriction and favor blood pressure raising [73]. This effect could be due to the impairment of NO basal vascular tone and endothelium-mediated vasodilation but above all by the SNS activation. This was found by Sakuma et al. [74] that after an acute intravenous administration of the NOS inhibitor L-NMMA, recorded an increase in BP, in norepinephrine values and in renal sympathetic nerve activity. They have also found a further increment of these responses after baroreceptor deafferentation while a disappearance was observed after cervical spine section, suggesting that L-NMMA pressor effects are mostly due to its effect on SNS. These results are supported by the evidence that ganglionic blockade and sympathectomy suppress BP and heart rate increase induced by another NOS inhibitor such as L-NAME [75, 76]. NO modulation is also particularly relevant in RVLM, the main bulbar area of integration of excitatory autonomic efferent fibers involved in cardiovascular regulation, and in hypothalamic paraventricular nuclei. Microinjection of NOS inhibitors at this level causes renal SNA and BP increase, while NO donors exhibit opposite effects [77, 78].
NO seems to play a role in the pathophysiology of diseases characterized by increased sympathetic discharge. A reduced activity of the intracerebral NO-pathway has been found in renovascular hypertension and heart failure [11, 79].
As regards to the neurogenic control of vascular function, it has been shown that sympathetically induced vasoconstriction may increase shear stress on the vascular wall with consequent increase in NO release from vascular endothelial cells favoring norepinephrine neuronal reuptake in sympathetic nerve terminals [80, 81].
Some other functions deserve to be mentioned. First, NOS may catalyze formation of hydrogen peroxide (H2O2) that is a potent vasodilator, but prolonged increased concentrations may be harmful to endothelial and vascular smooth muscle cells, leading to a shift in the balance between the production of protective NO and deleterious O2 − [82]. O2 − from the adventitia of the blood vessel can also inactivate NO [83]. It has been shown that oxygen-derived free radicals induced by low-density lipoproteins, Ang II, and pulsatile stretch in artery smooth muscle cells have been implicated in the pathogenesis of atherosclerosis and vascular restenosis in humans [84]. Second, NO has also the role to maintain a balance in the kidney between oxygen consumption and sodium reabsorption [85]. Third, NO released from the vascular endothelium plays an important role in the regulation of tissue mitochondrial respiration in skeletal muscle and in the regulation of cardiac contractile function [86, 87]. Fourth, in healthy adults, the pulmonary vascular resistance is maintained in part through the continuous local production of NO [88].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


