13 Neuroendocrine Lesions of the Lung
The concept of a “diffuse neuroendocrine system” (DNS) is not a new one. Feyrter1 developed this paradigm in 1938, in a philosophical attempt to unify tumors in several anatomic locations that had potential secretory functions and similar morphologic characteristics. Pearse2 refined and renamed the cellular network in question 35 years later, coining the designation of “APUD” system (for amine precursor uptake and decarboxylation) to describe its shared biochemical attributes. Inherent in the latter scheme was the presumption that all “APUD” cells—and tumors deriving from them (“APUDomas”)—emanated from the remnants of the neural crest. In light of these and other observations, continuing nosologic revisionism over the last 10 years has pushed many pathologists away from such traditional diagnostic terms as “carcinoid” and “islet cell tumor” in describing certain potentially malignant but low-grade neoplasms of the neuroendocrine system,3,4 although “traditionalists” remain.5 The classification of poorly differentiated lesions has changed as well. This chapter outlines the current foundations of existing classification schemes for neuroendocrine tumors. The reader should come away with a simplified—and therefore practical—understanding of this confusing area of oncology.
Terminology Pertaining to Neuroendocrine Neoplasms
There is perhaps no other single aspect of neuroendocrine neoplasia that is as perplexing as the pathologic terminology that has been used to describe it. Such terms as “bronchial adenoma,” “carcinoid,” “atypical carcinoid,” “Kulchitsky cell carcinoma,” “argentaffinoma,” “APUDoma,” “atypical endocrine carcinoma,” “oat cell carcinoma,” “medullary thyroid carcinoma,” “islet cell tumor,” and “cutaneous Merkel cell carcinoma” (group 1) have all been employed historically in this context, in addition to “neuroblastoma,” “esthesioneuroblastoma,” “olfactory neuroblastoma,” “medulloblastoma,” “pineoblastoma,” “retinoblastoma,” “paraganglioma,” “pheochromocytoma,” “chemodectoma,” and “glomus jugulare tumor” (group 2).6–10
This diverse lexicon reflects a basic division of neuroendocrine tumors into two broad categories—epithelial (group 1) and neural (group 2).6 With that piece of information in hand, one can then go on to structure a much more user-friendly and straightforward classification scheme that has made significant inroads in the pathology literature.
Another crucial concept in understanding the categorization and clinical behavior of neuroendocrine neoplasms is that all of them are at least potentially malignant tumors, regardless of whether they belong to group 1 or group 2.9 Moreover, in selected subgroups (e.g., classic “carcinoid” tumor, extra-adrenal paraganglioma [PG], and pheochromocytoma [intra-adrenal PG]), one cannot reliably use the gross or microscopic characteristics of the tumors to predict whether they will behave innocuously or aggressively. Therefore, it follows logically that the modifier “benign” should not be applied in conjunction with any of the diagnostic terms noted earlier. For example, even with regard to appendiceal or classic bronchial “carcinoids”—generally regarded as defining the “low” end of the spectrum of biologic behavior in this context11–13—there are many well-documented examples of metastasizing lesions that can be found in the literature.
Current terminological recommendations are, therefore, different from those that might have pertained even 10 years ago, or from those with which some practitioners may feel “comfortable.” The designation of “neuroendocrine carcinoma” (NEC) has been proposed as a replacement for all of the various group 1 terms discussed earlier, with modifiers of “well-differentiated” (grade I/III); “moderately differentiated” (grade II/III); and “poorly differentiated” (grade III/III) being appended as appropriate.9,12,14 In contrast, most of the traditional rubric has been retained with reference to group 2 tumors, such that the recommended terms for the categorization of this constellation of lesions are “extra-adrenal PG,” “intra-adrenal PG” (pheochromocytoma), “sympatheticoadrenal neuroblastoma (NB)” (and congeners [e.g., ganglioneuroblastoma]), “olfactory NB,” “retinoblastoma,” and “primitive neuroectodermal tumor” (PNET).9 In reporting on a biopsy or resection specimen, the pathologist can then work within this framework—using further descriptive comments and summaries of the aggregate literature on each tumor—to provide the clinician with an outline of expected behavior for each neoplasm based on its nuances.
Distribution and Pathogenesis of Neuroendocrine Neoplasia
If one refers to basic textbooks on human embryogenesis, a common theme that is seen in all anatomic sites is that of a neuroendocrine or neuroectodermal stage of differentiation during early organ development.6,15,16 Because it is currently thought that oncogenesis partially (and aberrantly) recapitulates normal embryologic development, this information is central to our understanding of why neuroendocrine and neuroectodermal neoplasms have been reported in virtually every topographic location. Some of the latter are, by far, more commonly hosts to such tumors, for unknown reasons. For example, the lung is the most common site of neuroendocrine carcinogenesis, where the process is clearly related etiologically to cigarette smoking and is associated with partial deletion of the short arm of chromosome 3.17 However, identical sporadic primary neoplasms in other organs have not been linked with any definitive pathogenetic factors.18 Group 2 neoplasms in the “peripheral primitive neuroectodermal” category similarly demonstrate a uniform balanced translocation between chromosomes 11 and 22,19,20 with synthesis of a unique gene product (p30/32 glycoprotein) that is recognized by a particular set of monoclonal antibodies (e.g., 12E7 and O13).20
Other NECs and group 2 tumors (especially NB and retinoblastoma) occur in definite Mendelian-heritable—typically autosomal dominant—patterns, as seen in multiple endocrine neoplasia (MEN) type 1 (pancreatic and thymic group 1 tumors) and MEN2 (medullary thyroid carcinoma and also group 2 tumors [pheochromocytomas]).9,11,21,22 Ongoing work has elucidated the locations of at least some operative aberrant gene complexes in such disorders (e.g., the MEN2A locus on chromosome 10 and deletion of the Rb-1 “anti-oncogene” on chromosome 13 in heritable bilateral retinoblastoma).17,19,23 However, sporadic examples of the tumors discussed earlier do not necessarily exhibit the same karyotypic or gene sequence abnormalities.24 Clearly, more work is needed to provide a complete picture of the molecular disturbances at play,14,25 but this is an exciting area for current and future development because it may yield clinical tests that could be used in early diagnosis and treatment.
Other Pathologic Aspects of Neuroendocrine Neoplasia
Up to this point, this discussion has focused on “pure” neuroendocrine and neuroectodermal neoplasms. Increasingly in recent years, however, it has been recognized that human malignancies much more often show combined or “divergent” differentiation than was appreciated in the past. Accordingly, oncologists are now faced with such diagnoses as “adenocarcinoma/squamous carcinoma/transitional cell carcinoma with neuroendocrine features” or, more simply, “combined adenocarcinoma–small cell NEC.”26–33 In the former scenario, the pathologist is attempting to convey the concept that the tumor looks like conventional squamous carcinoma, adenocarcinoma, or transitional cell carcinoma with a routine hematoxylin and eosin stain, but that additional studies (e.g., ultrastructural or immunohistochemical) have demonstrated submicroscopic neuroendocrine differentiation in the neoplastic cells. In some organ systems, such as the lung, it is believed that such a constellation of findings portends a more aggressive course of certain tumor types (e.g., “large cell anaplastic pulmonary carcinoma with neuroendocrine features”).34 However, generic extrapolation of this model to other tissues would not be scientifically justified at this time and appears to be definitely invalid in some specific settings.31,33 When a truly combined carcinoma is seen at a light microscopic level, pathologists are describing the juxtaposition and admixture of two distinct histologic patterns, such as adenocarcinoma and small cell carcinoma (SCC).29,30 Again, using tumors of the lung as examples, it would be expected that the responses to therapy and the behavior of such combined lesions would also be a hybrid of those attending each component (i.e., adenocarcinoma or SCC) in pure form.35–38 Nonetheless, uniform validation of that premise and delineation of optimal therapeutic approaches for each of these “amalgam” tumors have yet to occur.
Pathologic Recognition of Neuroendocrine Differentiation
Standard Morphologic Examination
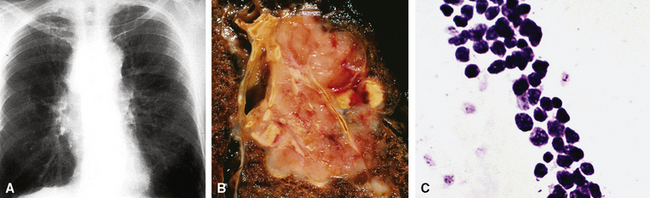
Because of the histologic appearance of some tumors, a neuroendocrine phenotype is obvious. Ready examples include classic pulmonary SCC and central bronchial “carcinoid” tumor. The standard cytomorphologic features of SCC are well known and include hyperchromatic nuclei with dispersed chromatin, inconspicuous nucleoli, and a tendency for the nuclei to mold to one another (Fig. 13-1). The cells are oval or carrot-shaped, with scant cytoplasm, and often there is marked crush artifact. Extensive necrosis is often observed, with either a geographic pattern or dropout of individual cells (i.e., apoptosis). Likewise, bronchial “carcinoid” features include stippled nuclear chromatin and a distinctly organoid growth pattern, with formation of rosettes, trabeculae, or ribbons (“festoons”)39 (Fig. 13-2).
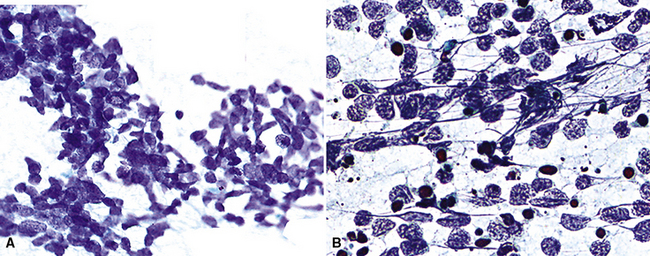
Histochemical Methods
Histochemical techniques are still valuable in selected settings, although in current practice, they have been supplanted by newer technologies. With reference to neuroendocrine lesions, such techniques rely on the ability of endocrine cells to reduce silver solutions and form insoluble precipitates in tissue sections. Available methods are broadly subdivided into two groups—argentaffin and argyrophil stains.40–42
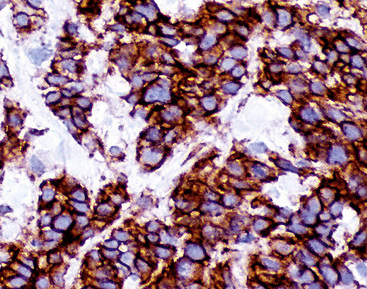
Argentaffin techniques depend on endogenous cellular reducing agents. The most widely used is the Fontana–Masson stain, which is only variably reactive with pulmonary neuroendocrine neoplasms. Argyrophilic methods are those in which an exogenous reducing agent is added, such as in the Grimelius or Churukian–Schenk procedure. They are typically positive in the majority of well-differentiated neuroendocrine tumors of the lung (Fig. 13-3), with much lesser reactivity in high-grade carcinomas, such as SCC.42
Electron Microscopy
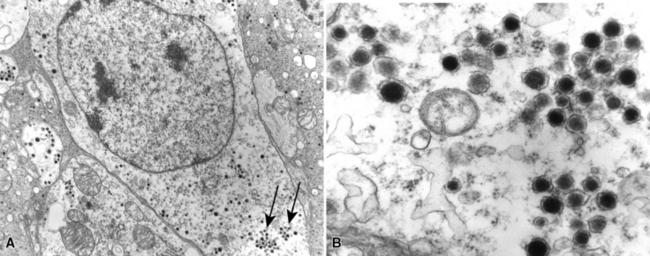
The ultrastructural hallmark of neuroendocrine neoplasms in the lung and elsewhere is the presence of cytoplasmic neurosecretory granules. These are rounded structures that consist of a central dense core, a peripheral lucent halo, and a single delimiting outer membrane. They vary from 30 to 300 nm in diameter, and may occur singly or in clusters42–55 (Fig. 13-4). Budding of neurosecretory granules from prominent Golgi apparatus is occasionally observed. One drawback of electron microscopy is that the number of granules and the number of cells containing granules tend to be greatest in well-differentiated tumors and lowest in poorly differentiated lesions (where they are most needed for diagnosis). Also, electron microscopy can examine only relatively few cells in any given tumor. However, it is still a highly useful technique if sufficient time and care are invested in a thorough search for dense core granules. In a study by Nagle and colleagues,40 ultrastructural analysis documented neuroendocrine differentiation in all of 41 putative neuroendocrine lesions, whereas a significant percentage of lesions were negative for endocrine markers using immunohistochemical techniques.
Immunohistology
Advances in immunohistochemistry over the last 15 years have yielded powerful tools for the pathologist in recognizing neuroendocrine differentiation.44,50,54–60 Advantages of this approach include a relatively low cost, rapid turnaround time, the ability to perform studies on routinely processed tissue, and the capacity to screen a large number of cells rapidly as opposed to the limited number that can be evaluated with electron microscopy. The following markers have the greatest utility in this context.
Intermediate Filament Proteins
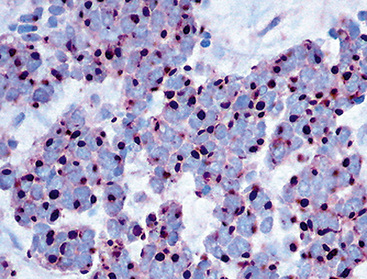
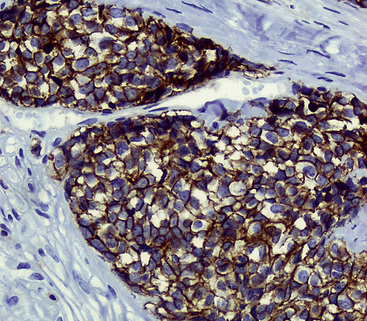
Group I neuroendocrine neoplasms manifest uniform immunoreactivity for keratin, especially keratin classes 8 and 18.61 To a lesser extent, keratin 19 is also observed in NECs in general. Pulmonary NECs may label for keratin 20 as well, but only in fewer than 10% of cases.62 Keratin proteins are generally well recognized by several commercial monoclonal antibodies, especially CAM5.2 and MFN116. Monospecific reagents directly against only one keratin class polypeptide are also available, but are not necessary in this specific context. In our opinion, the optimal approach to the detection of keratin in any poorly differentiated neoplasm, neuroendocrine or otherwise, is to prepare a mixture of monoclonal antikeratin antibodies with partially overlapping and partially distinctive keratin class specificities. When reagents of this type are used on paraffin sections, together with properly performed proteolytic digestion or microwave (heat)-mediated epitope “retrieval” methods, the detectability of keratin in NECs should approximate 100%, even in essentially “undifferentiated” tumors (e.g., SCC).63 Another feature of keratin reactivity in many NECs is a distinctive punctate or globoid perinuclear zone of positivity.64 This result (Fig. 13-5) is simultaneously diagnostic of both epithelial and neuroendocrine differentiation in a small cell malignancy, and it obviates the need for other “neuroendocrine markers.” Keratin is not typically expected in group II neuroendocrine neoplasms, although some lesions in that group have shown its presence in an “aberrant” manner.65–68 The foremost example of that phenomenon is represented by PNET with “divergent” differentiation (also known as “polyphenotypic small cell tumor”), an example of which is the “desmoplastic small round cell tumor.”69–71 In most cases of keratin expression in more nondescript PNETs, reactivity for that protein is focal, unlike its pattern in NECs; in addition, vimentin expression tends to be mutually exclusive in PNETs and NECs. Nonetheless, problems in differential diagnosis may arise between those classes of neuroendocrine tumors, sometimes necessitating cytogenetic evaluation to distinguish between them.72
Neurofilament protein is variably coexpressed by NECs,73–76 but that determinant is seen as the sole intermediate filament in the majority of differentiated group II neuroendocrine tumors, such as PGs and pheochromocytomas, as well as some NBs.77–79 Unfortunately, neurofilament protein is not well visualized in paraffin sections, even with epitope retrieval technology, and it is most reliably evaluated in frozen material. Vimentin also may be evident in the cells of PGs in approximately 50% of cases,79 and it is the only intermediate filament that can be detected in primitive group II tumors, such as NB and PNET.68 That is also true in Ewing sarcoma, a tumor that is in the same family as PNETs. Vimentin is only exceptionally present in NECs, as stated earlier. Eusebi and coworkers80 recently described three NECs that coexpressed keratin and desmin as a reflection of divergent (sarcomatoid) rhabdomyoblastic differentiation. This same proclivity is regularly seen in desmoplastic small round cell tumors, in which keratin, desmin, and vimentin are commonly present concomitantly in the same neoplastic cells.69–71
Glial fibrillary acidic protein is not an expected reactant in either NEC or PNET. In the central nervous system, therefore, this marker is helpful in the differential diagnosis with small cell malignant gliomas,81 most of which are reactive for glial fibrillary acidic protein. That fact is all the more important because a proportion of glial tumors manifest aberrant keratin reactivity82 and also express vimentin.
Chromogranins
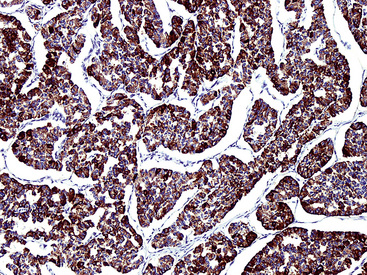
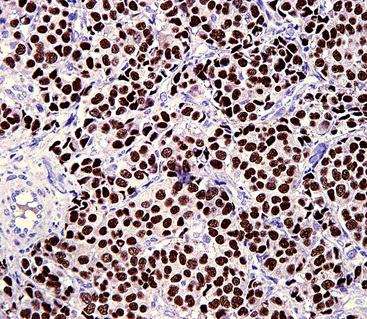
Chromogranins are matrical proteins that are associated with neurosecretory granules and are absolutely specific for neuroendocrine differentiation.83–85 These polypeptides have been subdivided biochemically into two groups, A and B,86,87 with the latter being synonymous with secretogranin-I. Conceptually, every cell that packages peptides into neurosecretory granules must synthesize either chromogranin-A (CGA) or chromogranin-B (CGB); hence, a screening reagent incorporating antibodies to both of those proteins would be extremely useful diagnostically. Unfortunately, reliable commercial anti-CGB products have not been introduced. The most widely used anti-CGA antibody is clone LK2H10.88 This reagent was raised against pheochromocytoma cells and shows excellent reactivity with paraffin sections (Fig. 13-6). The major shortcoming of anti-CGA is twofold. First, because neuroendocrine cells or tumors may preferentially synthesize CGB, CGA obviously cannot be regarded as a “universal” marker of such elements. Secondly, the detectability of both CGA and CGB is directly related to the number of neurosecretory granules in any given neuroendocrine cell population. If one is dealing with poorly differentiated malignancies that have only scant numbers of such organelles, immunostains for CGA and CGB will be predictably negative in the majority of cases. Therefore, one may legitimately conclude that a tumor has neuroendocrine properties if it can be labeled for a chromogranin, but negative results do not exclude that possibility, particularly in high-grade neoplasms.
Synaptophysin
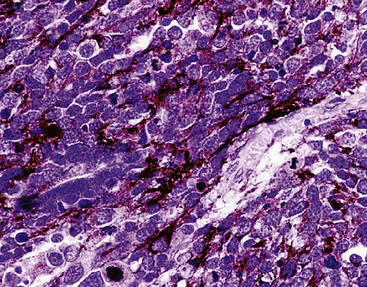
Synaptophysin is a 38 kDa molecule that is associated with the synaptic vesicles of neurons and cells with neuroendocrine or neuroectodermal characteristics.78,89–92 Monoclonal antibodies to this marker have been used widely in diagnostic surgical pathology and cytopathology, with good success, particularly in conjunction with epitope retrieval techniques (Fig. 13-7). In light of its subcellular associations, one might assume that synaptophysin would have a synonymous tissue distribution to that of the chromogranins; however, in practicality, that is not true. Thus, a sizable proportion of neuroendocrine neoplasms label for CGA or CGB, but not synaptophysin; the converse also applies.93 Thus, anti-synaptophysins and anti-chromogranins should be conceptualized as complementary reagents.
Loy and colleagues59 have challenged the specificity of chromogranins and synaptophysin as markers of neuroendocrine lineage, in a study using ultrastructure as the standard for definition of that lineage. However, in our opinion, the concept behind that analysis was flawed. Sampling bias (a significant problem in fine structural evaluations) probably accounted for those cases in which immunoreactivity was observed for the specified endocrine markers, but no neurosecretory granules were identified by electron microscopy.
CD57 (Leu-7)
CD57 was originally characterized as a marker for natural killer lymphocytes, and it is recognized by monoclonal antibody HNK-1,94 among others. Subsequently, shared epitopes of this molecule have been detected immunohistochemically in normal constituents and tumors of the brain, peripheral nervous system, soft tissues, prostate gland, thymus, and DNS.95–101 With specific reference to neuroendocrine neoplasms, CD57 appears to bind to a matrical component of neurosecretory granules that is distinct from both CGA and CGB.102 It both corroborates and extends the sensitivity of chromogranin immunostains in many clinical settings. Nonetheless, because of the imperfect specificity of Leu-7, it cannot be used as confidently as a “standalone” neuroendocrine marker. Moreover, it has the same failings in sensitivity as CGA and CGB that relate to the density of neurosecretory granules in poorly differentiated tumors.
Neural Cell Adhesion Molecule (NCAM; CD56)
The monoclonal antibodies 123C3 and JLP5B9 recognize formalin-preserved epitopes of CD56, or neural cell adhesion molecule, a cell membrane protein that has a role in the cohesiveness of cells in the peripheral and central nervous systems.103–109 Like synaptophysin, NCAM is distributed among neuroendocrine and neuroectodermal cells and tumors (Fig. 13-8). Several studies have shown that NCAM is a sensitive marker of endocrine lineage in SCC of the lung as well as extrapulmonary sites.106–108 Nonetheless, it is not absolutely specific for neuroendocrine differentiation; a minority (up to 25%) of ovarian surface carcinomas, renal cell carcinomas, nonendocrine lung cancers, and endometrial carcinomas demonstrate CD56 immunoreactivity.108 Despite this caveat, antibodies to NCAM are useful additions to diagnostic panels for endocrine tumors. Reagents raised against the polysialylated form of CD56 are considered the most sensitive among this group.105
Neuron-Specific (Gamma-Dimer) Enolase
“Neuron-specific” enolase (NSE) is a gamma-dimeric form of 14-3-2 protein, a glycolytic enzyme that is present in neurons and cells of the DNS.110 It is thought to be associated with the formation of intercellular synapses in the nervous system. NSE was one of the first markers to be used in diagnostic immunohistochemistry as a general indicator of putative neuroendocrine differentiation.110–112 Heteroantisera to NSE are very sensitive in that regard, labeling virtually all neuronal and neuroendocrine proliferations, regardless of the level of cellular differentiation.112 Nevertheless, the specificity of those reagents is poor, owing to cross-reactivity between gamma-dimers and heterodimers (e.g., alpha-gamma, alpha-beta) of NSE that are expressed by non-neuroendocrine neoplasms of many types.113 In reaction to that problem, monoclonal antibodies to NSE have been developed and tested,114 but these have generally manifested a low degree of sensitivity and have not enjoyed widespread usage. An alternative approach to lessening the cross-reactivity of anti-NSE heteroantisera is to absorb them with acetone-fixed, pulverized human tissues known to contain high levels of the alpha or beta form of the molecule. However, that approach is time-consuming and somewhat demanding technically, and for this reason, has not been embraced in clinical practice. Currently, antibodies to NSE are primarily used as “screens” for endocrine differentiation,115–117 and reactivity with them is usually pursued further by applying other neuroendocrine immunostains, as described elsewhere in this chapter.
Protein Gene Product 9.5 (PGP9.5)
PGP9.5 is a protein that removes ubiquitin (an intermediate filament) from other proteins and protects them from degradation by proteases. It is expressed widely within cells and tumors of the DNS and therefore has been applied as an immunohistochemical marker of neuroendocrine differentiation.118–120 Nevertheless, the general distribution of PGP9.5 in human neoplasms has shown that it is not particularly specific for an endocrine lineage; in particular, non–small cell carcinomas of the lung that lack neuroendocrine morphologic patterns and contain no neurosecretory granules on ultrastructural analysis have been PGP9.5-positive in approximately 55% of cases in some series.121 Thus, the practical utility of this marker is similar to that of NSE.
Specific Neuropeptides
Specific neuropeptides, such as adrenocorticotropic hormone, gastrin, insulin, somatostatin, calcitonin, “whole” bombesin and its C-terminal flanking peptide, leu- and met- enkephalins, were the initial molecular moieties that were evaluated immunohistochemically in an effort to substantiate the neuroendocrine nature of selected epithelial human neoplasms.93 Today, these markers have been largely relegated to a secondary role in the pathologic assessment of tumors in the DNS. Antibodies to such peptides are of academic interest in correlating clinical endocrinopathies with histopathologic findings, but are not nearly sensitive enough to serve as screening reagents. Another potential but limited application is in the delineation of “occult” (non–endocrinopathy-related) peptide production by neuroendocrine tumors, which may serve as the basis for serologic monitoring of tumor growth and activity.
CD99 (MIC2; p30/32 Protein)
The membranocytoplasmic protein known as “CD99” in the hematopoietic antigen cluster designation is the same molecule that has been called “MIC2” or “p30/32 protein.”122 The function of this moiety is uncertain, but it is empirically known to be expressed in virtually all PNETs and Ewing sarcomas122,123 (Fig. 13-9). The specificity of CD99 antibodies for a neuroectodermal lineage is not absolute, however, because they may also label a minority (<15%) of alveolar rhabdomyosarcomas as well as the overwhelming majority (90%) of lymphoblastic lymphomas.124 CD99 is observed in up to 20% of NECs in some body sites as well.125,126 This is important because it may obscure the difference between NEC and PNET in selected instances; this is particularly true in light of the potential for keratin reactivity that both lesions have. On the other hand, differentiated group II neuroendocrine tumors, such as PGs, are nonreactive for this determinant.
Other Reagents
Several other antibodies have been developed that have immunohistochemical properties that overlap those of LK2H10 (anti-CGA). These include both polyclonal and hybridoma reagents. A monoclonal antibody designated “HISL-19” by Krisch and coworkers127 is believed to manifest neuroendocrine specificity. The determinant that it recognizes is proteinaceous, but seems to be nonidentical to chromogranin. This statement is based on the relative strength of reactivity of HISL-19 with several neuroendocrine tissues, which is dissimilar to that of LK2H10. Moreover, some nonendocrine tissues and tumors, such as the gallbladder epithelium and adenocarcinomas of the lung, stomach, and endometrium, are labeled by the former reagent but not by the latter. These differences notwithstanding, HISL-19 appears to bind to a neurosecretory granule-related moiety, and neoplasms with few of these granules (parathyroid adenoma, melanoma, NB, and oat cell lung cancer) are only weakly stained, but others with numerous granules (pituitary adenoma, pheochromocytoma, medullary thyroid carcinoma, carcinoid, and islet cell tumor) are labeled intensely. Western immunoblot analyses have shown that the target of HISL-19 is not neuron-specific enolase.127
Lloyd and colleagues128 employed monoclonal antibodies to adrenaline (epinephrine) and noradrenaline (norepinephrine) in the study of similar neuroendocrine neoplasms. In general, pheochromocytomas displayed adrenaline alone, but extra-adrenal PGs exhibited reactivity for adrenaline and noradrenaline, or noradrenaline only. NBs, carcinoids, pituitary adenomas, pancreatic endocrine tumors, and parathyroid adenomas also showed positivity for noradrenaline. These authors emphasized that catecholamine-positive neoplasms also expressed reactivity with LK2H10, implying that stains for adrenaline and noradrenaline did not add appreciably to the diagnostic information obtained with antichromogranin.
Haspel and colleagues129 found that mice infected with reovirus type I often had autoimmune syndromes featuring polyendocrinopathies. Spleen cells from such animals were used as the substrates for hybridoma production, and two of the resulting monoclonal antibodies (5B5 and 5B8) showed reactivity with human anterior pituitary cells in frozen and Bouin’s-fixed specimens.129 Further immunostaining results in normal and neoplastic human tissues were not provided, but it was believed that 5B5 and 5B8 recognized discrete hormonal products, such as growth hormone, based on competitive inhibition studies.
The absence of discussion of S-100 protein may seem surprising because of the still-common belief that this marker is associated with neuroendocrine tumors. In reality, that is not so. The only endocrine neoplasm that reproducibly contains cells positive for S-100 protein is PG, and the immunoreactive elements therein are actually “sustentacular” cells rather than tumor cells.79 It has been contended that sustentacular elements decrease in density when PGs undergo malignant change,130 but that is an uncertain tenet in reference to individual cases.
Several markers have been assessed with regard to their associations with neuroendocrine tumor grade; SOX2, PAX5, and CD117 are the principal analytes. Each demonstrates a tendency for greater reactivity in this context as the histologic grade increases, such that grade III lesions show the highest level of immunolabeling.131–133
Yet another applicable reagent is antithyroid transcription factor-1 (TTF1).134–140 This intranuclear protein is expressed not only by thyroid epithelium but also by glandular cells of the lung, pulmonary adenocarcinomas, and NECs of the lung (Fig. 13-10). Among the last of these lesional groups, SCC and large cell NECs have the highest incidence of reactivity (in approximately 85% to 90% of cases). Interestingly, however, extrapulmonary high-grade NECs show a tendency to be TTF1-positive in many organ sites (bladder, uterine cervix, prostate, gastrointestinal tract), and therefore this marker can be considered to represent an “adjunct” neuroendocrine determinant, if additional studies for thyroid-related and lung-associated labels are negative. Obviously, then, TTF1 cannot be employed reliably to distinguish metastatic NEC of the lung from other neuroendocrine malignancies.
Specific Features of Pulmonary Neuroendocrine Proliferations
Before further discussion of specific neuroendocrine proliferations in the lung, a general consideration of the current nosology is in order. Travis et al.141,142 recently provided a classification scheme for pulmonary neoplasms in general through the auspices of the World Health Organization (WHO; Box 13-1).
Box 13-1 World Health Organization Classification of Pulmonary Neuroendocrine Lesions
From Travis WD, Colby TV, Corrin B, et al: Histological Typing of Lung & Pleural Tumours: International Histological Classification of Tumours. Geneva: World Health Organization; 1999:1–55.
As discussed earlier, we believe that all neuroendocrine proliferations of the lung are at least potentially malignant. The only exceptions are pulmonary tumorlets and neuroepithelial bodies, which are considered hyperplastic rather than neoplastic. As others have done,143 we espoused the scheme shown in Box 13-2 as more appropriate than the WHO paradigm in the general categorization of neuroendocrine lesions.
Box 13-2 Alternative Classification of Neuroendocrine Proliferations of the Lung
The following points constitute our rationale for this type of classification:
At this time, we make the diagnosis of “NEC,” followed by its grade, for all type I (epithelial) neuroendocrine tumors of the lung.144 Parenthetically, the traditional terminology is usually used as well to facilitate transitions in nomenclature.145 At some point, it should be possible to omit the older terminology altogether.
Grade I Neuroendocrine Carcinoma (“Classic Carcinoid”)
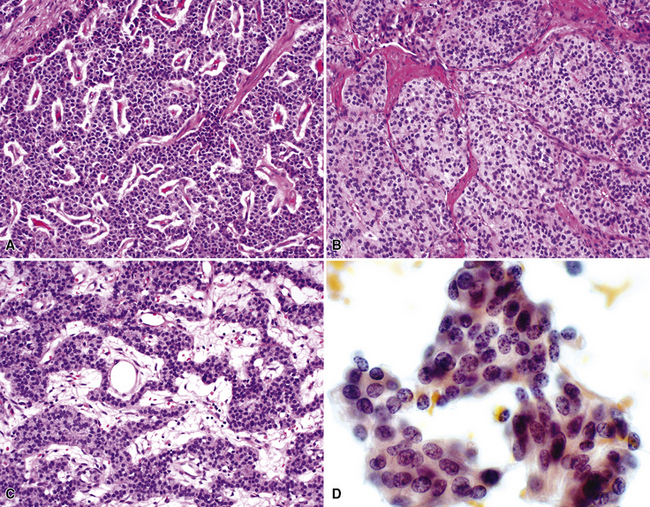
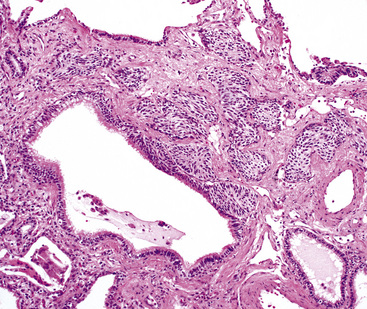
Bronchopulmonary “carcinoid” (grade I NEC) was initially considered an “adenoma” of the bronchus,146 a term that unfortunately persists in the lexicon of some individuals. The similarity of this lesion to gastrointestinal carcinoids, or “carcinoma-like tumors,” described 30 years earlier, was noted. Although the majority of classic carcinoids are centrally located, approximately 10% to 20% are found in the periphery (Fig. 13-11) of the lung.11,12,15,45,147–152 An anatomic landmark that is commonly used to distinguish central and peripheral tumors is the cartilaginous airways; neoplasms associated with such structures are considered central, and others without that relationship are considered peripheral.148
Clinical Features
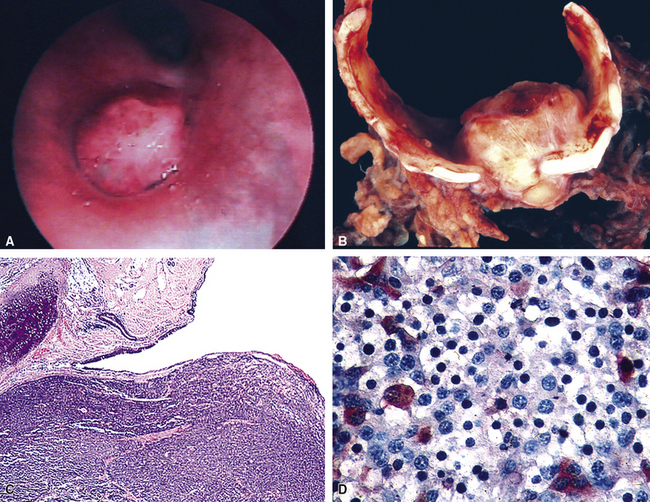
Grade I NECs with typical histologic features are rarely diagnostic problems. They usually grow as polypoid intraluminal masses with an intact overlying epithelium (Fig. 13-12) or one demonstrating squamous metaplasia.45,147,148 This pattern explains a common clinical presentation, which is localized airway obstruction. Patients manifest with wheezing, cough, or pneumonia. Carcinoid syndrome almost never develops, presumably because the tumors in question are incapable of synthesizing the biochemical substances responsible for its pathogenesis. Rare individuals with grade I NEC have associated Cushing syndrome or other endocrinopathies as a result of ectopic neuropeptide production by the neoplasm (see Fig. 13-12D).153–159 A proposed association between pulmonary carcinoids and sarcoidosis has also been made.160
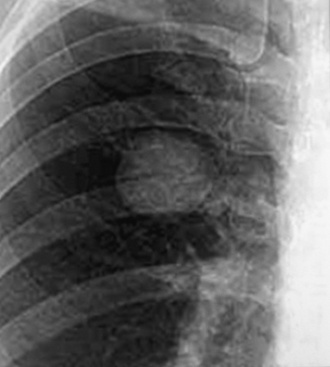
Localized obstruction also dominates the radiographic picture, with evidence of obstructive pneumonia or atelectasis (Fig. 13-13). Occasionally, a central mass with a dumbbell-like configuration is seen on plain films, and computed tomography usually demonstrates a lesion within and adjacent to a large airway.161
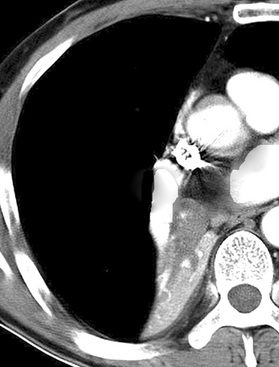
Peripheral carcinoids are subpleural, small, and well circumscribed.148,151,152 They often present as incidental findings, lacking the propensity to produce the obstructive changes of their central counterparts. Instead, the differential diagnosis of a “coin lesion” is encountered. Radiographically, such entities as granulomas, hamartomas, or peripheral adenocarcinomas enter consideration. Some studies have shown a predominance of peripheral grade I NECs in the right middle lobe, and a greater number of women have those tumors compared with central NECs of the lung.151,152
Rarely, grade I NECs may be synchronously multifocal.162,163 That eventuality is troublesome because of the possibility of metastasis to the lung from an extrapulmonary source. Extensive clinical evaluations and appropriate immunohistologic studies (discussed later) are usually required.
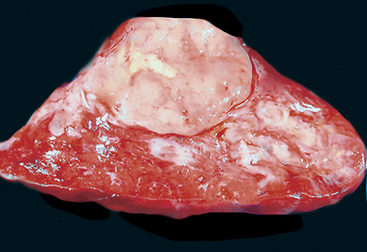
Gross and Microscopic Pathologic Findings
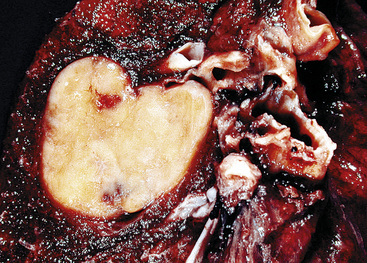
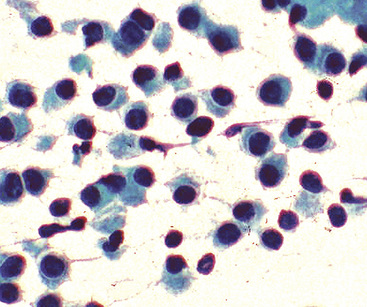
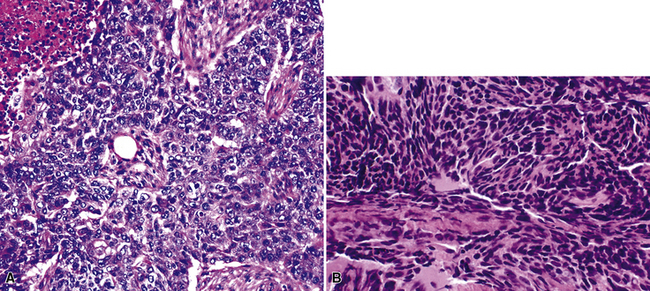
“Classic carcinoids” are typically 2 to 4 cm in maximum dimension. The lesions vary in color from tan-yellow to dark red, and they lack obvious internal necrosis and hemorrhage. Central tumors display diverse growth patterns, including trabecular, ribboning, insular, and solid sheet-like configurations11,147,148 (Fig. 13-14). A delicate fibrovascular stroma is present, sometimes with associated matrical amyloid deposition.164 Rare lesions may contain metaplastic bone or cartilage.11 The latter findings are postulated to reflect the production of factors related to tumor growth factor beta or various bone morphogenetic proteins.148 Tumor cell cytoplasm is relatively abundant, and it may be strikingly granular and oncocytic or even clear.165–169 Eccentricity of the nuclei can yield a plasmacytoid cellular image, particularly in fine-needle aspiration biopsy specimens (Fig. 13-15). A papillary configuration has also been reported.11,170 Other variants of grade I NEC produce melanin,171,172 and some are said to contain sustentacular cells immunoreactive to S-100 protein, as is more typically seen in PGs.173 An exceptional case presented by Skinner and Ewen174 featured diffuse replacement of the lung parenchyma by carcinoid tumor cells.
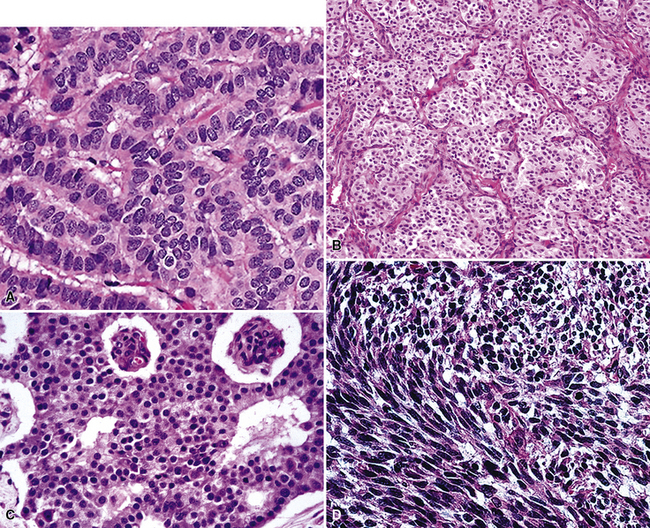
Peripheral carcinoids may be morphologically identical to central tumors, in which case the diagnosis is usually made without difficulty. Nonetheless, peripheral grade I NECs often demonstrate a prominent spindle cell growth pattern, which is associated with a somewhat different set of diagnostic alternatives (discussed later)151,152,175 (Fig. 13-16).
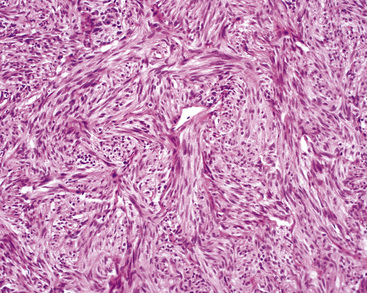
Differential Diagnostic Considerations
The intraluminal growth pattern of grade I pulmonary NEC can be simulated by salivary gland-like tumors, such as mucoepidermoid carcinoma; mesenchymal lesions, including peripheral nerve sheath tumors and rare inflammatory pseudotumors; and some metastatic neoplasms. Most of these possibilities present little difficulty histologically. Nonetheless, problems may arise occasionally in differentiating carcinoids from well-differentiated adenocarcinomas, higher-grade NECs, primary pulmonary PGs, and “solid” adenoid cystic carcinomas, particularly in small transbronchial biopsy specimens, which may demonstrate significant artifactual distortion. Distinction from “atypical” carcinoids is discussed in more detail later. However, uniform nuclear features, the lack of significant mitotic activity, abundant cytoplasm, and the absence of necrosis are all indicative of grade I NEC rather than a grade II tumor.3,5,149 Because of the well-differentiated nature of classic carcinoids, one can expect almost all of them to be diffusely reactive for keratin, CGA, synaptophysin, and CD57,4,173 whereas the other diagnostic alternatives do not show that immunophenotypic profile. The p53 gene, as studied by immunohistochemical or genetic analysis, is infrequently mutated in grade I NEC,176 as opposed to higher-grade neuroendocrine tumors.177
Another differential diagnostic consideration in this specific context is metastatic grade I neuroendocrine NEC from a source outside the lungs. Srivastava and Hornick178 immunohistologically compared pulmonary with nonpulmonary well-differentiated NECs, finding that the presence of thyroid transcription factor 1 was restricted to primary tumors of the lung. Classic pulmonary carcinoids also lacked reactivity for CDX2 and PDX1, both of which are alimentary tract–related markers.
Spindle cell growth in grade I NECs raises a dissimilar set of differential diagnostic considerations. These include fibrous and smooth muscle neoplasms, fibrous pseudotumors, primary or metastatic spindle cell melanomas, and metastatic medullary thyroid carcinomas. Careful attention to nuclear detail in spindle cell carcinoids shows retention of the typical stippled chromatin pattern, and nucleoli are small and inconspicuous. The lesions also express the majority of immunohistologic markers seen in central carcinoids, as discussed earlier. A combination of morphologic and adjunctive studies should be capable of excluding most of the differential diagnostic considerations mentioned earlier, but metastatic medullary thyroid carcinoma is virtually indistinguishable from grade I pulmonary NEC on pathologic grounds.164 Likewise, Eyden and coworkers179 have shown that metastatic melanomas sometimes exhibit partial neuroendocrine differentiation in ultrastructural or immunohistochemical studies.
A similar issue is the separation of tumorlets (Fig. 13-17) from peripheral carcinoids. This is an arbitrary distinction because immunohistochemical and ultrastructural studies indicate that the cells of these two lesions are essentially identical.180–182 Also, there is debate over whether tumorlets represent true neoplasms or are instead hyperplasias of respiratory neuroendocrine cells.15,181,183 They generally occur within and around small bronchioles, and they typically have a spindle cell composition. The small nests of tumor cells in a neuroendocrine tumorlet tend to be divided by a fibrous stroma into small packets, and this feature helps to differentiate neuroendocrine tumorlets from small peripheral carcinoid tumors, where tumor cell growth is more sheetlike, with variable fibrous stands interlaced. Although such tumorlets, which may number in the hundreds in some cases, are said to occur most often in diseased lungs with such associated lesions as bronchiectasis or pulmonary fibrosis,180–184 they also may arise in otherwise normal lungs.162 Rizvi and colleagues185 and Ferolla and colleagues186 have also shown that neuroendocrine hyperplasia is much more common in patients with pulmonary NECs than in others with non-neuroendocrine neoplasms. In any event, an arbitrary size of 4 mm or less has been suggested for tumorlets; larger lesions are considered peripheral carcinoids.181 A continuum for such proliferations was suggested by Miller and Muller,187 who found that peripheral carcinoid tumors were often associated with diffuse neuroendocrine cell hyperplasia and airway obstruction. A particular pitfall associated with tumorlets is that they may be misdiagnosed as much more aggressive lesions in small biopsy specimens, especially when clinical data are ignored or unavailable.188,189
Treatment and Outcome
The clinical outcome in cases of central grade I NECs of the lung is generally excellent.5,11,45 Complete excision is the treatment of choice, possibly necessitating lobectomy or sleeve resection.190 Conservative endobronchial excision is generally associated with an unacceptable rate of local recurrence.190–193
A peripheral location or a spindle cell growth pattern does not, in and of itself, equate with “atypical” morphology in grade I NECs of the lung. The prognosis for low-grade spindle cell peripheral lesions is equivalent to that of their central relatives, despite a slightly higher rate of lymph node metastasis in some series.45,151,152
Figures for the incidence of metastasis differ widely and depend on two major variables. These include the definition of peribronchial lymph node metastasis (i.e., direct invasion of nodes vs. embolic metastatic involvement) and the histologic purity of the primary lesion. Some studies with high rates of metastasis for classic “carcinoid” have improperly included higher-grade neuroendocrine lesions. Quoted metastasis rates vary from 1% to 20%; a figure of approximately 5% to 10% is probably closest to the actual incidence, and most secondary implants involve adjacent peribronchial or hilar lymph nodes.5,192–196 It is this behavioral attribute that leads us, as well as others, to consider central pulmonary carcinoids irrefutable carcinomas.143 Distant metastases also occur rarely, and there is a tendency for spread to the bones, liver, skin, or brain.45,193,194 Interestingly, osseous metastases are typically blastic.197 Flow cytometric measurement of DNA content in grade I NECs is not helpful in predicting their metastatic potential.149,198
The overall survival rate for patients with grade I NEC of the lung is greater than 95% at 5 years.5,45,148,192–196,199 Metastatic disease may be palliated with interleukin or somatostatin analogs,38,200 and surgical excision of secondary implants can be considered, given the slow growth of this tumor type.
Grade II Neuroendocrine Carcinoma (“Atypical Carcinoid”)
The term “atypical carcinoid” was first used by Arrigoni and coworkers in 1972.201 Those authors reviewed 216 bronchial carcinoids treated at the Mayo Clinic and found 23 with unusual features, including pleomorphism, mitotic activity, nuclear hyperchromatism with increased nucleocytoplasmic ratios, and evidence of spontaneous necrosis or hemorrhage. In the original series, 70% of the lesions metastasized and seven patients (30%) died of their tumors. Several terms have subsequently entered the literature on this lesion, including “Kulschitzky cell carcinoma”202 and “well-differentiated NEC,”12,15,194,202–209 and the criteria for their definition are still being debated.3,5,35
Clinical Findings
“Atypical carcinoids” of the lung are usually greater than 3 cm in maximum dimension. Other characteristics include an etiologic link to cigarette smoking, differing from the epidemiologic features of grade I NECs.5,201–216 Grade II NECs are more likely to be peripherally located in the lung (Fig. 13-18) compared with “classic” carcinoids; the former lesions have been so situated in the majority of cases in several studies.201–216 Potential associations with Cushing syndrome217 and Eaton-Lambert syndrome also apply to these lesions.
Pathologic Findings
With the exception of the possible gross identification of hemorrhage or necrosis and a tendency to be slightly larger, the lesions were difficult, if not impossible, to distinguish from typical carcinoids by clinical, radiographic, or gross pathologic evaluation (Fig. 13-19). The pathologic requirements for a diagnosis of “atypical carcinoid” have varied from study to study, but most have included mitotic activity and necrosis. We currently use the criteria of El-Naggar and colleagues149 to define grade II NEC of the lung (Fig. 13-20), preferring that term to “atypical carcinoid,” but stipulating that the two are conceptually synonymous. Those requirements center on the following points:
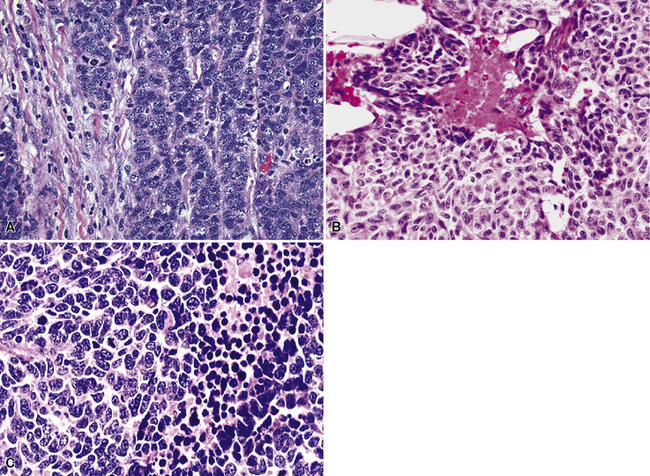
Spindle cell change in grade II NECs of the lung is potentially seen but uncommon.215 As observed by Yousem and Taylor,3,218 a requirement for at least two of these criteria is more reasonable than basing the diagnosis of grade II NEC on only one feature in a lesion that is otherwise a classic carcinoid of the lung.
Immunohistochemical studies show some differences between grade I NEC and grade II NEC of the lung. As expected of more poorly differentiated tumors, “atypical carcinoids” tend to express fewer markers of neuroendocrine differentiation or to demonstrate more focal labeling for them; such markers as synaptophysin, CGA, and CD57 are usually less impressive in grade II lesions.92,117,120,219,220 Reactivity for specific neuropeptides—such as bombesin, calcitonin, and adrenocorticotropic hormone (ACTH)—is relatively uncommon.221 Mutant p53 protein is demonstrable by immunostaining in some atypical carcinoids, as opposed to its absence in the great majority of typical carcinoids but like its presence in most SCCs.176,222,223
Ultrastructural analysis of grade II NEC shows fewer neurosecretory granules compared with grade I neuroendocrine neoplasms.207 Cytogenetic analysis also has demonstrated multiple karyotypic abnormalities in grade II NEC but not in classic carcinoid,224–227 paralleling the flow cytometric incidences of aneuploidy in the two tumor types.149,218,228
Differential Diagnosis
In addition to its distinction from other neuroendocrine lesions, the differential diagnosis of grade II NEC includes poorly differentiated non-neuroendocrine carcinomas of the lung.44,50,53,210 In particular, selected examples of high-grade adenocarcinoma may simulate the microscopic configuration of “atypical carcinoids,” and “basaloid carcinoma” (basaloid squamous cell carcinoma)229 is regularly confused with grade II NEC. The nuclear characteristics of those lesions differ from one another in that non-neuroendocrine tumors do not manifest the dispersed chromatin seen in “atypical carcinoids,” instead showing more vesicular nuclei with often-prominent nucleoli (Fig. 13-21). Immunohistochemical studies may assist with the differential diagnosis in this context, but because some grade II NECs lack reactivity for endocrine markers and conversely selected non-neuroendocrine carcinomas of the lung demonstrate their presence, this technique is problematic. Electron microscopy is still probably the surest technique for differentiating grade II NEC from its non-neuroendocrine diagnostic simulators.
Treatment and Clinical Outcome
A vexing problem is which diagnostic label to assign a neuroendocrine pulmonary tumor with only one indicator of potentially aggressive behavior, such as angiolymphatic invasion, brisk mitotic activity, aneuploidy, or large size. None of these features independently appears to warrant adjuvant therapy, but it is probably prudent to suggest that they may be associated with an adverse outcome and to monitor the patient carefully for recurrence. Special techniques may provide additional information. In a flow cytometric analysis by El-Naggar and colleagues,149 approximately 80% of “atypical carcinoids” were aneuploid, whereas 80% of classic carcinoids were diploid. Also, grade II lesions were more likely to have an S-phase fraction of greater than 7%. Both of these factors were statistically linked to a worse outcome. However, in that analysis, the most significant predictor was accurate morphologic separation into grade I and grade II categories. Rush and colleagues,206 Jackson-York and coworkers,228 and Travis and colleagues230 concluded that accurate classification of these tumors is more important in the prognosis than DNA content. Other factors said to significantly decrease survival rates in cases of “atypical carcinoid” include lymph node metastases, vascular invasion, and overall tumor size of greater than 3 cm.5,148,201–209,210–216,230
Lymph node metastases are found at diagnosis in 30% to 50% of cases, and approximately 25% of patients with grade II NEC of the lung have remote metastatic disease at presentation.5,215,216,231 As in cases of SCC, the brain is a common site for metastasis and recurrence. Importantly, the mortality rate for this neoplasm is 30% to 50% at 2 years.5,148,201–209,210–215 Rush and colleagues206 have reported 5- and 10-year survival rates of 60% and 40%, respectively. Obviously, these figures reflect a different biologic potential than those of classic carcinoid and SCC. The use of adjunctive therapy in the management of grade II NEC of the lung is unresolved.232,233 Some researchers have suggested that chemotherapy, irradiation, or both can be beneficial in this setting, but there has been no consensus on that point, much less on which pharmacologic agents to use. The difficulty in securing diagnostically “pure” study populations for treatment evaluations has likely contributed to this uncertainty.
Grade III Neuroendocrine Carcinoma, Small Cell Type
Small cell carcinoma is probably the best recognized neuroendocrine neoplasm in this discussion, accounting for approximately 25% of all lung carcinomas.32 That diagnostic entity is generally traced to Barnard’s 1926 description of “oat cell carcinoma”234; although the tumor had been believed to represent a lymphoma or sarcoma before that time, he accurately recognized its epithelial nature. In 1959, Azzopardi refined the pathologic description of SCC.235
Clinical Features
Small cell carcinoma of the lung has many potential modes of clinical presentation. These include symptoms and signs similar to those of other common carcinomas of the lung, such as cough, hemoptysis, weight loss, anorexia, fatigue, and the syndrome of hypertrophic osteoarthropathy.236,237 In addition, however, unusual findings, such as hyponatremia, hypercalcemia, or Cushing syndrome, may emanate from ectopic production by the tumor of antidiuretic hormone, parathyroid hormone-like peptides, and ACTH, respectively.155,159,236,238,239 Rarely, individuals with SCC manifest Eaton-Lambert (pseudomyasthenia) syndrome owing to paraneoplastic interference with neuromuscular function.240,241 Retinopathy, central and peripheral neuropathies, encephalitis, glomerulonephritis, cutaneous reactions, sarcoid-like granulomatous lesions, and systemic vasculitis also have been reported in this context.241–248 Massive hepatomegaly with liver failure,249 myelophthisic anemia,250,251 or seizure activity and headaches252 are additional clinical constellations related to the effects of distant visceral metastases at initial diagnosis.
Chest radiographic findings may be relatively unremarkable or may demonstrate a central hilar (or, more rarely, a peripheral intrapulmonary) mass (Fig. 13-22). Obstructive pneumonia is a potential complication; bulky peribronchial or mediastinal lymphadenopathy is common and may be massive.161,236 Pleural effusions typically signify serosal metastases of SCC, and rarely, the tumor may so markedly involve the pleura that it produces a peripulmonary “rind” similar to that seen in association with mesotheliomas.253