Figure 5–1.
The interaction between neural triggers and cardiovascular substrate during autonomic activation [2]. Stimulation of beta1-adrenergic receptors can decrease electrical stability directly as a result of changes in second messenger formation and alterations in ion fluxes. This deleterious influence is opposed by muscarinic receptor stimulation, which inhibits presynaptically the release of norepinephrine and opposes its action at the receptor level. Catecholamines may also alter myocardial perfusion by complex means, including alpha-receptor stimulation of coronary vessels and platelets and by impairing diastolic perfusion time due to adrenergically mediated sinus tachycardia (From Verrier [2]. Reprinted with permission from Springer Dordrecht)
Useful noninvasive tools have been developed to explore the influence of autonomic factors on arrhythmogenesis, including heart rate variability [3], baroreceptor sensitivity [3], heart rate turbulence [4], and deceleration capacity [5]. The direct effects of neural stimuli on the electrical instability of the myocardial substrate can be assessed by monitoring repolarization indices, notably T-wave alternans, measured by either the Spectral or Modified Moving Average (MMA) Methods [6].
Integration of Neural Control of Cardiac Electrical Activity
Regulation of cardiac neural activity is highly integrated and is achieved by circuitry at multiple levels (Fig. 5.2) [7]. Higher brain centers operate through elaborate pathways within the hypothalamus and medullary cardiovascular regulatory sites. Baroreceptor mechanisms have long been recognized as integral to autonomic control of the cardiovascular system, as evidenced by heart rate variability and baroreceptor sensitivity testing of both cardiac patients and normal subjects. The intrinsic cardiac nerves and fat pads provide local neural coordination independent of extrinsic cardiac nerves and higher brain centers. Newly recognized is the phenomenon of electrical remodeling attributable to nerve growth and degeneration. At the level of the myocardial cell, autonomic receptors influence G proteins to control ionic channels, pumps, and exchangers. Finally, studies of behavioral state provide evidence that markers of arrhythmia vulnerability can be monitored noninvasively in combination with autonomic parameters during emotional and physical stressors and sleep states to identify individuals at heightened risk of lethal cardiac arrhythmias.
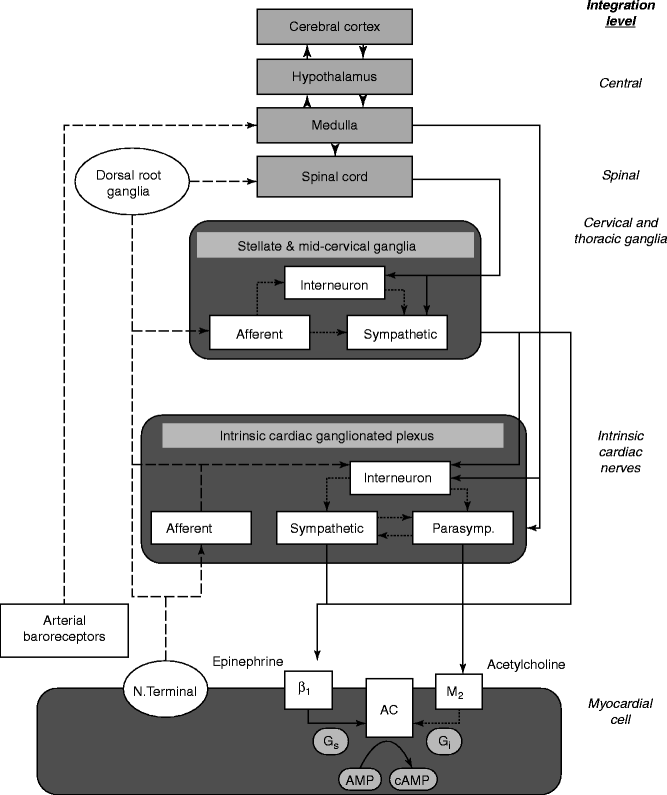
Figure 5–2.
Synthesis of new and present views on levels of integration important in neural control of cardiac electrical activity [7]. More traditional concepts focused on afferent tracts (dashed lines) arising from myocardial nerve terminals and reflex receptors (e.g., baroreceptors) that are integrated centrally within hypothalamic and medullary cardiostimulatory and cardioinhibitory brain centers and on central modulation of sympathetic and parasympathetic outflow (solid lines) with little intermediary processing at the level of the spinal cord and within cervical and thoracic ganglia. More recent views incorporate additional levels of intricate processing within the extraspinal cervical and thoracic ganglia and within the cardiac ganglionic plexus, where recently described interneurons are envisioned to provide new levels of noncentral integration. Release of neurotransmitters from postganglionic sympathetic neurons is believed to enhance excitation in the sinoatrial node and myocardial cells through norepinephrine binding to beta1-receptors, which enhances adenyl cyclase (AC) activity through intermediary stimulatory G-proteins (G s ). Increased parasympathectomy outflow enhances postganglionic release and binding of acetylcholine to muscarinic (M 2 ) receptors, and through coupled inhibitory G-proteins (G i ), inhibits cyclic AMP production (cAMP). The latter alters electrogenesis and pacemaking activity by affecting the activity of specific membrane Na, K, and Ca channels. New levels of integration are shown superimposed on previous views and are emphasized here to highlight new possibilities for intervention (From Lathrop and Spooner [7]. Reprinted with permission from John Wiley and Sons)
T-Wave Alternans as a Tool for Noninvasive Assessment of Neurocardiac Interactions
TWA, defined as a repeating ABAB pattern in the morphology and amplitude of the ST-segment or T wave, has been widely used in experimental and clinical studies to assess neural influences on susceptibility to cardiac arrhythmias. Furthermore, this parameter has proved to be useful in stratification of arrhythmia risk in prospective clinical studies enrolling >12,000 patients, as recently reviewed in a clinical consensus guidelines statement [6]. The mechanistic basis for TWA’s predictivity has been extensively discussed [8–10]. TWA appears to reflect spatiotemporal heterogeneity of repolarization, is sensitive to perturbations in intracellular calcium handling, and serves as a mechanism of arrhythmogenesis by amplifying repolarization heterogeneity. Diverse physiologic factors influence TWA, including heart rate, neurotransmitters, myocardial ischemia, and heart failure. The changes in TWA magnitude correlate with the pro- and antiarrhythmic effects of diverse interventions including pharmacologic agents [11]. The clinical presentation of TWA and its relationship to sudden death risk is illustrated in Fig. 5.3 [12, 13].
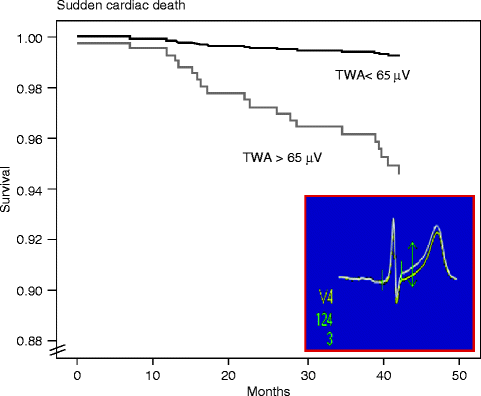
Figure 5–3.
Survival curves from the Finnish Cardiovascular Study (FINCAVAS), which enrolled >1,000 consecutive patients referred for routine exercise testing [12]. Inset is a high-resolution QRS-aligned template from a FINCAVAS patient illustrating T-wave alternans (TWA) as the separation between successive beats as measured by the Modified Moving Average method [13] (From Nieminen et al. [12]. Reprinted with permission from Oxford University Press. Inset from Minkkinen et al. [13]. Reprinted with permission from John Wiley and Sons)
Adrenergic Influences on Cardiac Vulnerability
It is well established that adrenergic inputs constitute the primary neural trigger for ventricular arrhythmias. Activation of the sympathetic nerve structures, including the posterior hypothalamus or stellate ganglia, increases susceptibility to ventricular fibrillation. Infusion of epinephrine or norepinephrine is also profibrillatory. Proof of the triggering role of sympathetic nerve discharge in ventricular arrhythmias was provided by Zhou et al., who performed direct recording of left stellate ganglion in ambulatory dogs with chronic myocardial infarction to demonstrate that ventricular tachycardia and sudden death are immediately preceded by spontaneous sympathetic nerve discharge (Fig. 5.4) [14]. Such striking surges in sympathetic nerve activity also occur within a few minutes of experimental left anterior descending (LAD) coronary artery occlusion and are associated with a marked increase in susceptibility to ventricular fibrillation, as evidenced by a fall in ventricular fibrillation threshold (Fig. 5.5) [15], as well as by spontaneous occurrence of the arrhythmia and correlated increase in TWA magnitude [16, 17]. Upon reperfusion, a second peak in ventricular arrhythmia vulnerability and TWA occurs, probably due to washout products of cellular ischemia [15–18]. Stellectomy significantly blunts the surge in vulnerability to ventricular fibrillation during occlusion but enhances its magnitude during reperfusion [16]. These findings are consistent with the facts that adrenergic factors play a key role during ischemia [19] and that stellectomy increases the reactive hyperemic response to release-reperfusion, which in turn probably leads to greater liberation of ischemic byproducts.
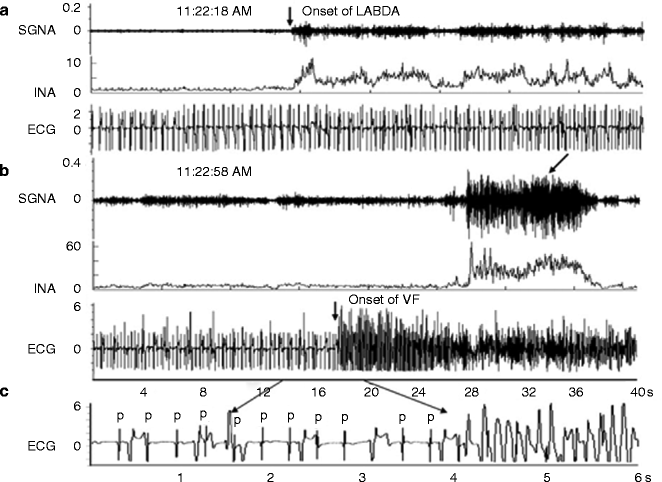
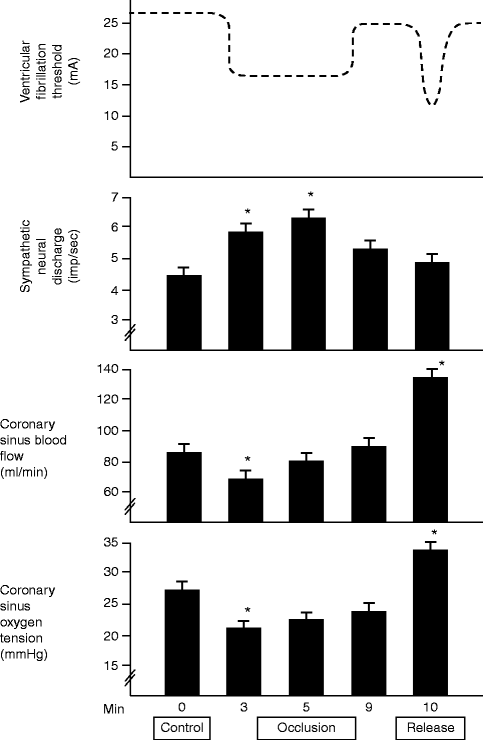
Figure 5–4.
Example of increased left stellate ganglion nerve activity (SGNA) preceding ventricular fibrillation (VF) and sudden cardiac death [14]. (a) Increased low-amplitude burst discharge activity (LABDA) resulted in accelerated idioventricular rhythm. (b) VF occurred approximately 40 s later. Panels a and b are continuous. (c) A 6-s recording from panel b. INA integrated nerve activity, P P wave, which is dissociated from ventricular activation due to complete AV block. Units for integrated nerve activity are millivolts (From Zhou et al. [14]. Reprinted with permission from Elsevier)
Figure 5–5.
Effects of a 10-min period of left anterior descending (LAD) coronary artery occlusion and release on neural sympathetic activity, coronary sinus blood flow, and oxygen tension [15]. A schematic representation of the time course of changes in ventricular fibrillation threshold is also displayed. LAD coronary artery occlusion results in a consistent activation of sympathetic preganglionic fibers, which corresponds with the period of maximal increase in vulnerability to ventricular fibrillation (* = p < 0.05 compared to control period). The concomitant changes in coronary sinus blood flow and reperfusion are also displayed (From Lombardi et al. [15]. Reprinted with permission from Elsevier)
In the atrium, simultaneous sympathovagal activation facilitates the onset of paroxysmal atrial fibrillation [20, 21] by a mechanism termed “calcium transient triggering.” Sympathetic nerve activation prolongs intracellular calcium transients, and vagus nerve activation shortens cardiac action potentials. The discrepancy between the normally tightly coupled action potential duration and calcium transients leads to increased forward Na/Ca exchanger current, which contributes to the generation of early afterdepolarizations towards the end of phase 3 of the cardiac action potential [22]. These triggers are particularly common in highly innervated pulmonary veins [23, 24].
Mechanisms Responsible for Arrhythmogenesis During Beta-Adrenergic Receptor Activation
The mechanisms whereby enhanced sympathetic nerve activity increases cardiac vulnerability in the normal and ischemic heart are complex. The major indirect effects include impairment of oxygen supply–demand ratio due to increased cardiac metabolic activity, alpha-adrenergically mediated coronary vasoconstriction, especially in vessels with damaged endothelium, and changes in preload and afterload. The direct arrhythmogenic effects on cardiac electrophysiologic function, which are primarily mediated through beta1-adrenergic receptors, are multifold. They include derangements in impulse formation, conduction, repolarization alternans, and heterogeneity of repolarization, with the potential for culmination in ventricular tachycardia and fibrillation (Fig. 5.6) [25, 26]. Increased levels of catecholamines stimulate beta-adrenergic receptors, which in turn alter adenylate cyclase activity and intracellular calcium flux. These effects are probably mediated by the cyclic nucleotide and protein kinase regulatory cascade, which can alter spatial heterogeneity of calcium transients and consequently provoke TWA and dispersion of repolarization. The effect of increased intracellular calcium, with the potential for overload and impaired intracellular calcium cycling by the sarcoplasmic reticulum may be compounded and become especially arrhythmogenic during concurrent myocardial ischemia, which further predisposes to intracellular calcium excess [16, 26–28]. The net effect is an increase in vulnerability to ventricular fibrillation. The converse is also true: reduction of cardiac sympathetic neural drive by stellectomy provides an antifibrillatory influence in animals and humans.
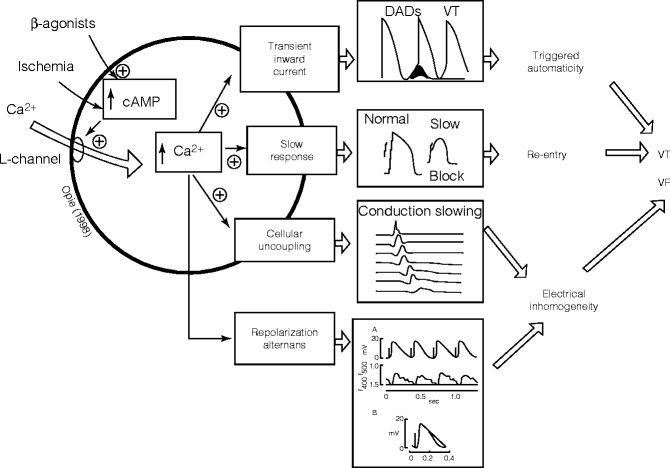
Figure 5–6.
The cardiac beta-adrenergic signaling system mediating ventricular arrhythmogenesis. The central pathways include links between cyclic adenosine 3′,5′-monophosphate (cAMP), cytosolic calcium, and specific calcium-mediated electrophysiologic abnormalities that predispose to ventricular tachycardia (VT) and ventricular fibrillation (VF). The lowest panel is based on a study from Lee et al. [26], indicating that simulated ischemia results in alternation in calcium transients, which appears to underlie action potential alternans (Adapted from Opie [25] and used with his permission; Lee [26] figure reprinted with permission from Wolters Kluwer Health)
Cardiac beta1-adrenergic receptor blockade is capable of negating the profibrillatory effect of direct sympathetic nerve stimulation [29] by an action at the neurocardiac effector junction. But, cardiac beta2-adrenergic receptors do not appear to play a significant role in modulating ventricular excitable properties. The role of cardiac beta3-adrenergic receptors has been enigmatic. Beta3 receptors are present in human cardiac myocytes in smaller quantities and are activated at higher concentrations of catecholamines than are beta1 and beta2 receptors. Zhou and coworkers [30] have provided evidence that during conditions of sympathetic hyperinnervation, there is a significant and dynamic response in beta3-adrenoreceptor expression. They also found that beta3 receptor stimulation in canines following myocardial infarction reduces the incidence of ventricular arrhythmias [31], suggesting that beta3 receptors serve a rescue function in response to catecholamine excess not only with respect to mechanical contractility (Fig. 5.7) [32, 33] but also to arrhythmia vulnerability. The mechanisms by which beta3 receptor agonism are antiarrhythmic is the subject of further investigation.
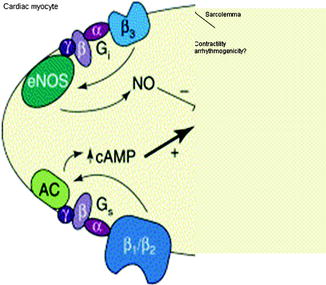
Figure 5–7.
Countervailing mechanisms proposed by Gauthier and coworkers [32] to account for the interaction between beta1– and beta2-adrenoceptors and the beta3-adrenoceptor subtype. In normal heart, beta1– and beta2-adrenoceptors mediate the classic positive inotropic effect of catecholamines via cAMP. In an opposing manner, stimulation of the beta3-adrenoceptor exerts a negative inotropic response that occurs through activation of a constitutively expressed endothelial nitric oxide synthase (eNOS). They propose that the beta3-adrenoceptor can provide a “rescue” function, which occurs particularly in disease conditions associated with hyperadrenergic activity, especially in heart failure. Little is known about the influence of these receptors on susceptibility to arrhythmias (Adapted from Gauthier et al. [32] by Verrier [33] and reprinted with permission from Elsevier)
Alpha-Adrenergic Receptors
Elucidation of the role of alpha-adrenergic receptors has been challenging because these agents exert direct actions not only on myocardial excitable properties but also on platelet aggregability and coronary hemodynamic function [34, 35]. In the normal heart, alpha-adrenergic receptor stimulation or blockade does not appear to affect ventricular electrical stability, as evidenced by the fact that administration of alpha-adrenergic agonists such as phenylephrine or methoxamine does not influence excitable properties when the pressor response is controlled to prevent reflex changes in autonomic tone [36, 37]. In the setting of myocardial ischemia, alpha-adrenergic blockade may alleviate coronary vasoconstriction and reduce platelet aggregability.
Sympathetic-Parasympathetic Interactions
Vagal influences are contingent on the prevailing level of adrenergic tone [38–42]. When sympathetic tone to the heart is augmented by thoracotomy [39], sympathetic nerve stimulation [39], myocardial ischemia, or catecholamine infusion [41], vagal activation exerts a protective effect on ventricular vulnerability. Vagus nerve stimulation alone is without effect on ventricular vulnerability when adrenergic input to the heart is ablated by beta-adrenergic blockade [39]. Levy and coworkers termed this phenomenon “accentuated antagonism.” The basis for this antagonism of adrenergic effects is presynaptic inhibition of norepinephrine release from nerve endings [43] and a muscarinically mediated action at the second messenger level, attenuating the response to catecholamines at receptor sites. Also, importantly, vagal influences provide indirect protection against ventricular fibrillation by reducing excess heart rates [39], which can otherwise critically compromise diastolic perfusion time during acute myocardial ischemia to increase ischemic insult. However, the beneficial effects of vagus nerve activity may be annulled if profound bradycardia and hypotension ensue. Vagus nerve stimulation has been shown in experimental studies to protect against ventricular arrhythmias during myocardial ischemia but its protection during reperfusion is attributable to decreased heart rate [44]. Finally, myocardial infarction may damage nerve pathways, thereby limiting the potential of the vagus nerve to be activated. Vanoli and colleagues demonstrated the antifibrillatory effect of vagus nerve stimulation during exercise-induced ischemia in canines with a healed myocardial infarction [45]. Direct stimulation of the right cervical vagus through a chronically implanted electrode at 15 s after onset of exercise-induced acute myocardial ischemia reduced the incidence of ventricular fibrillation by 92 %. This effect was only partly due to the attendant heart rate reduction, as in half of the animals, the efficacy of vagal stimulation persisted despite maintenance of constant heart rate by atrial pacing.
In the atrium, simultaneous activation of the sympathetic and parasympathetic nervous systems is thought to be highly conducive to atrial arrhythmias, due to synergistic effects on action potential characteristics, refractoriness, and propensity for triggered activity [20, 21]. This critical interaction between sympathovagal activation and cellular electrophysiology is most pronounced at sites of high concentrations of autonomic nerves at pulmonary vein-atrial junctions [24, 46]. In clinical studies using heart rate variability parameters, it was estimated that 10–25 % of atrial arrhythmias are facilitated by sympathovagal nerve influences, as atrial fibrillation was immediately preceded by a sharp increase in vagus nerve tone occurring on a background of increased sympathetic nerve tone [47]. Experimentally, in vitro infusion of acetylcholine in the sinus node artery invariably induced atrial fibrillation in dogs; acetylcholine-mediated atrial fibrillation was facilitated by the β(beta)-adrenoceptor agonist isoproterenol, which decreased the threshold concentration of acetylcholine required for arrhythmia induction and also prolonged episodes of atrial fibrillation [48].
Baroreflexes and Arrhythmias
The classic studies by Billman et al. [49] drew attention to the importance of baroreceptor function on susceptibility to life-threatening arrhythmias associated with myocardial ischemia and infarction. In their initial investigations in canines, they demonstrated that the more powerful was the baroreflex response, the less vulnerable animals were to ventricular fibrillation during myocardial ischemia superimposed on prior myocardial infarction. The protective effect of the baroreceptor mechanism has been linked primarily to the antifibrillatory influence of vagus nerve activity. The latter effect improves diastolic coronary perfusion, minimizing the ischemic insult from coronary artery occlusion. The importance of baroreceptor sensitivity (BRS) was subsequently documented in human subjects in whom baroreceptor function was evaluated with the pressor agent phenylephrine. LaRovere and colleagues [3] demonstrated that post-myocardial infarction patients were less likely to experience sudden cardiac death if their baroreceptor function was not depressed.
Experimental evidence indicates that exercise training improves depressed BRS in high-risk post-MI dogs and prevents VF during acute myocardial ischemia [50] and that it also provides antifibrillatory protection in high-risk dogs with a normal heart [51]. These findings paved the way for clinical studies to assess whether increasing vagal activity by exercise training is capable of significantly improving long-term prognosis. Ninety-five post-myocardial infarction patients, matched for all major variables, were randomized to a 4-week endurance-training period or to no training. During a 10-year follow-up, cardiac mortality among the trained patients who had an exercise-induced increase in baroreflex sensitivity >3 ms/mmHg was strikingly lower compared to that of the trained patients without such a baroreflex response and to that of the non-trained patients (Fig. 5.8) [52].
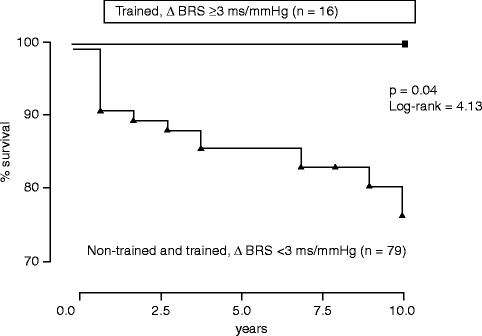
Figure 5–8.
Cardiac mortality estimated by the Kaplan-Meier method among the patients with a training-induced increase in BRS ≥3 ms/mmHg and the group including patients who trained without the same BRS increase and non-trained patients [52] (From La Rovere et al. [52]. Reprinted with permission from Wolters Kluwer Health)
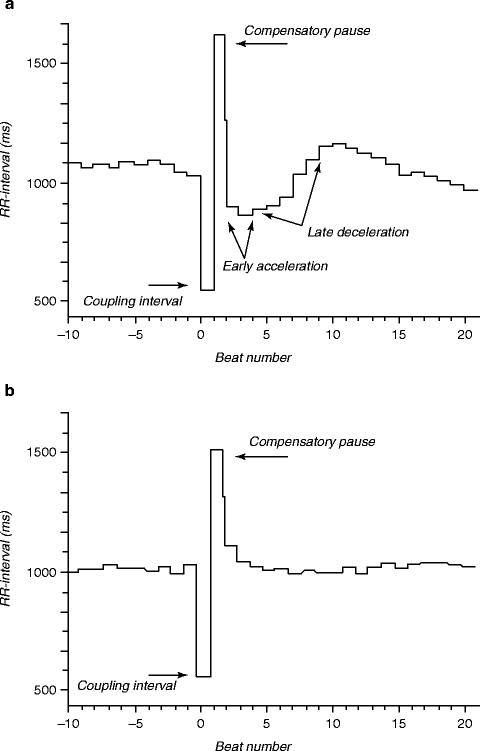
In the last few years, BRS testing has been pursued by noninvasive monitoring of heart rate turbulence (HRT) [4]. This phenomenon refers to fluctuations of sinus-rhythm cycle length after a single ventricular premature beat (VPB) and appears to be mechanistically linked with BRS [53]. The basic principle, introduced by Schmidt and coworkers, is that the reaction of the cardiovascular system to a VPB and the subsequent decrease in arterial blood pressure is a direct function of baroreceptor responsiveness, since reflex activation of the vagus nerve controls the pattern of sinus rhythm (Fig. 5.9) [54]. Several studies confirm that in low-risk patients, after a VPB, sinus rhythm exhibits a characteristic pattern of early acceleration and subsequent deceleration. By contrast, patients at high risk exhibit essentially a flat, nonvarying response to the VPB, indicating inability to activate vagal nerves and their cardioprotective effect. The method appears to be a promising independent predictor of total mortality in patients with ischemic heart disease, myocardial infarction, and/or heart failure [54–56]. Heart rate deceleration capacity, a related and even more comprehensive marker of autonomic control than HRT, may be of considerable clinical value in assessing overall autonomic regulation of the heart in patients with diverse types of cardiovascular disease [5].
Intrinsic Cardiac Innervation
In the late 1970s, Armour [57] and his colleagues introduced and investigated the elaborate intrinsic neural network within the heart, which provides local, independent heart rhythm control. Randall, Zipes, Chen and their respective coworkers [58–61] subsequently verified this important advance, drawing attention to the fact that components of this innervation system reside within discrete fat pads. Myocardial ischemia can compromise the functional capacity of cardiac intrinsic neurons residing in the fat pad and thus has the potential to increase electrical inhomogeneity and susceptibility to arrhythmias [62]. Intrinsic innervation is also vulnerable to diabetic neuropathy, which accordingly could exacerbate vulnerability to arrhythmias [63]. Surgical incisions through the atrial walls and radiofrequency ablation may isolate SA node pacemaker cells and damage the fat pads and result in proarrhythmia due to iatrogenically induced autonomic imbalance [64]. Heterogeneity of fibers within and without the fat pads contributes to dispersion of electrical activity, which in turn can predispose to arrhythmogenesis in adjacent atrial tissue [65]. Finally, another region that is richly innervated by the autonomic nervous system is the pulmonary vein-atrial junction, which contains a high density of co-localized sympathetic and parasympathetic ganglionated plexi from which atrial fibrillation may frequently arise [46].
Nerve Growth and Degeneration
Whereas the concept of remodeling has been well established with respect to the heart, the importance of restructuring of cardiac innervation has only recently received due attention, with fundamental contributions from the laboratories of Zipes [66, 67] and Chen [68–71]. In particular, Jayachandran et al. [67] demonstrated in a canine model of atrial fibrillation induced by rapid, prolonged pacing, that atrial electrical remodeling was associated with spatially heterogeneous uptake of the postganglionic sympathetic indicator hydroxyephedrine into the nerve terminals within the sinus node, crista terminalis, and myocardium. Importantly, increased uptake was accompanied by electrical heterogeneity and augmented norepinephrine tissue levels. Studies by Chang et al. [69] and Olgin et al. [66] provided further evidence in favor of the concept of injury-induced neural repair with selective sympathetic remodeling and the attendant potential for induction and perpetuation of atrial arrhythmias.
Chen and coworkers [70] documented evidence that nerve sprouting could apply to ventricular as well as to atrial arrhythmogenesis and potentially to sudden cardiac death. These investigators demonstrated a significant correlation between increased sympathetic nerve density as reflected in immunocytochemical markers and history of ischemia in native hearts of human transplant recipients. In a canine model, they determined that induction of nerve sprouting with nerve growth factor resulted in increased incidence of ventricular tachycardias and sudden death, with concomitant TWA [71], consistent with this parameter’s capacity to track arrhythmia vulnerability [6]. Significantly, the predisposition to arrhythmias was linked to immunocytochemical evidence of a heterogeneous pattern of sympathetic nerve reinnervation (Fig. 5.10) [72]. More recently, Liu and coworkers [73] demonstrated in rabbits that hypercholesterolemia can produce neural and electrophysiological remodeling that is highly arrhythmogenic and is associated with important changes in ionic currents including ICa. Collectively, this evidence points to the lability of autonomic innervation and the intricate changes that may be responsible for derangements in neural activity. This adverse effect of heterogeneous remodeling of sympathetic innervation to the heart is likely to play a role in the increased risk for life-threatening arrhythmias [72]. The term “neural remodeling” should be employed alongside “myocardial remodeling” in the conceptual framework of the pathophysiology of acute infarction.
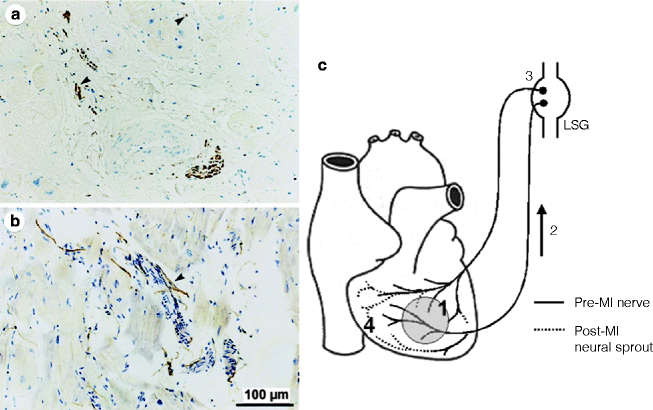
Figure 5–10.
Nerve sprouting after myocardial infarction. Panels a and b demonstrate TH-positive nerve fibers (arrowheads) in injured areas or around coronary arteries and in a patient with coronary artery disease [68] (From Cao et al. [68]. Reprinted with permission from Wolters Kluwer Health). Panel c Signaling of neural remodeling after myocardial infarction [72]. Myocardial injury (shaded area) results in early local nerve growth factor (NGF) release, presumably from damaged cells, followed by upregulated NGF and growth-associated protein 43 (GAP43) expression, especially in the infarct area (1). These signal proteins are then retrogradely transported (2) to the nerve cell bodies in the ganglia (3) where they stimulate the sprouting of new cardiac nerve endings in the heart (4), predominantly in noninfarcted regions, leading to heterogeneous hyperinnervation (From Verrier and Kwaku [72]. Reprinted with permission from Wolters Kluwer Health)
Behavioral State
Stress and Arrhythmogenesis
Behavioral models have been developed to define the impact of behavioral state on cardiac electrical stability [28, 74–76]. These have included both aversive behavioral conditioning paradigms and models eliciting natural emotions, notably anger and fear. Aversive conditioning of dogs in a Pavlovian sling with mild chest shock on three consecutive days showed that subsequent exposure to the environment without shock elicited a reduction in the repetitive extrasystole threshold >30 % [74]. The same paradigm elicited a threefold increase in the occurrence of spontaneous ventricular fibrillation when coronary artery occlusion was carried out in the aversive sling compared to the nonaversive cage environment. In dogs recovering from myocardial infarction, exposure to the aversive environment consistently elicited ventricular tachycardia for several days during the healing process [75]. After this time, the animals continued to exhibit signs of behavioral stress in the aversive environment but no longer experienced ventricular arrhythmias, indicating that the arousal state required a substrate of cardiac electrical instability for the induction of rhythm disturbances. The stress-induced changes in cardiac excitable properties were largely obtunded by beta-adrenergic receptor blockade with propranolol or metoprolol.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


