Figure 13–1.
High amplitude spike discharge activity (HASDA) induced ventricular tachycardia (VT) (Panel a), premature ventricular contraction (PVC, arrow) (Panel b) and QRST morphology change (arrows, Panel c) in dogs with MI, atrioventricular block and NGF infusion to the left stellate ganglion (From Zhou et al. [21] with permission)
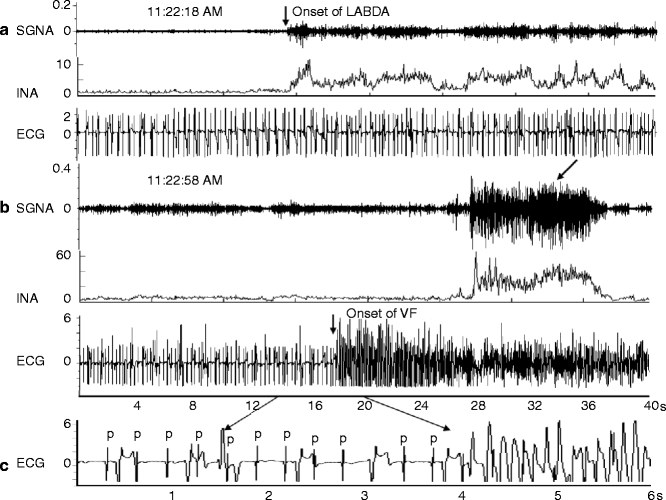
Figure 13–2.
An example of increased left stellate ganglion nerve activity (SGNA) preceding VF and SCD. Panel a shows increased LABDA resulted in accelerated idioventricular rhythm. Panel b shows that VF occurred approximately 40 s later. Panels a and b are continuous. Panel c shows a view of 6 s recording from panel b. P, P wave, which is dissociated from the ventricular activation due to complete atrioventricular block. The arrow on ECG tracing points to the onset of VF. The arrows on the SGNA tracing point to massive sympathetic discharges after the onset of VF. INA integrated nerve activity. The unit for INA in this and all other figures is μV (From Zhou et al. [21] with permission)
Defibrillation and Intracellular Ca2+ Dynamics
As shown in Fig. 13.2, there is increased sympathetic discharges both preceding and following the onset of VF. The sympathetic nerve activity releases norepinephrine, which increases the intracellular Ca2+. Abnormally large Ca2+ transient is known to be a mechanism of cardiac arrhythmia [22]. Zoll et al. [1] showed that electrical defibrillation is effective in terminating VF. However, in the same manuscript, the authors showed VF may also recur spontaneously soon after electrical defibrillation. The mechanisms by which electrical shocks terminate VF remain unclear, so are the mechanisms by which VF may spontaneously recur after VF-defibrillation episodes. Since the strong electrical shocks alter the transmembrane potentials by creating a voltage gradient field, defibrillation was thought to be achieved exclusively by the changes of membrane potential through electrical current. However, recent experimental data have demonstrated that defibrillation shocks also change Cai, which may be of great importance to the outcome of the defibrillation.
Mechanisms of Defibrillation
To understand the mechanisms of defibrillation, it is necessary to also understand the mechanisms of defibrillation failure. The mechanisms of failed defibrillation depend on the strength of defibrillation shocks [23, 24]. At weak strength shocks (typically >100 V lower than the shock strength with 50 % probability of successful defibrillation (DFT50)), reentrant activation immediately after the shock contributes to the defibrillation failure by maintaining continuous VF. Optical mapping studies have reported that there was no post-shock interval at weak shock strengths and more phase singularities remained after delivery of the weaker shocks [23, 24]. To achieve defibrillation, the virtual electrode polarizations (VEPs) created by the shock-induced electric field need to have sufficient amplitude to halt the activation wavefronts of VF. However, this is not the only reason of defibrillation failure at weak shock strengths. Even if the VEPs extinguish all existing VF activation wavefronts, it is not sufficient to ensure successful defibrillation, because the VEPs can themselves generate new activation wavefronts that reinitiate VF. Since VF consists of unsynchronized rapid activations, some portions of the ventricular myocardium are in their vulnerable period no matter when the shock is delivered. When a critical value of the potential gradient encounters a critical degree of refractoriness of the tissue during the vulnerable period, reentry is formed at the critical point, which reinitiates VF [25, 26]. Stronger electric shocks prevent the formation of the critical point and the re-initiation of VF, because of the presence of an upper limit to the strength of shocks given during the vulnerable period that could induce VF (i.e. the upper limit of vulnerability) [27, 28]. Another type of the critical point has been reported in a study by Efimov et al. [29]. In this pattern, the critical point is created during the action potential plateau by shock-induced VEPs of depolarization and hyperpolarization. The hyperpolarization caused by virtual anode de-excites the myocardium during the plateau of the action potential, leading to recovery of the excitability at the hyperpolarized region. This allows the adjacent depolarized region caused by virtual cathode to activate the hyperpolarized region, giving rise to a new activation wavefront and initiation of reentry (i.e. the virtual electrode-induced phase singularity) [29]. Electric shocks at a sufficiently strong strength also prevent the virtual electrode-induced phase singularity, because an increase in the shock strength enhances the degree of depolarization and hyperpolarization at the virtual cathode and anode, respectively, which accelerates the conduction velocity of the activation wavefront across the boundary of each VEP [30]. The electric shock can evoke an action potential in the excitable segment of the ventricular myocardium during VF. However, the weak strength shocks can exert little effects on the myocardium during a refractory phase of the action potential, which leave the ventricular repolarization unsynchronized. Therefore, the critical point of either mechanism induced by electric shocks can induce reentry in the tissue with heterogeneous repolarization at weak shock strengths.
On the other hand, the mechanism of defibrillation failure differs at stronger shock strengths near defibrillation threshold (DFT) (shock strength within 50 V of DFT50). It has been reported that the near DFT strength shock applied during the refractory period is capable of extending the APD and the effective refractory period [31]. The degree of the APD extension depends on the shock coupling interval, thereby causing the myocardium to repolarize at a constant time regardless of the pattern of the pre-shock electrical activities [32]. This synchronized repolarization elicited by the strong electric shock prevents the genesis of the shock-induced critical point and reentry. The optical mapping study has identified that the number of post-shock phase singularities decreases continuously from pre-shock values to zero as the shock strength increases [24]. Importantly, even though reentry was not observed immediately after the near DFT strength shock, defibrillation does not necessarily succeed. In other words, immediate post-shock reentry is not responsible for defibrillation failure at near DFT shock strengths. In that case, activation is not recorded in both electrical and optical mapping studies for a period of 50–90 ms following the near DFT shocks (i.e. the isoelectric window), after which earliest activation always propagates centrifugally away from the early site in a focal pattern [23, 33–35]. If the shock fails to defibrillate, these foci arise rapidly and repetitively for several cycles, resulting that the activation wavefronts degenerates back into VF. If the shock successfully defibrillates, no foci are observed or only a few focal activations last until they stop spontaneously. One question is whether the shock extinguishes all VF activations during isoelectric window. Many electrical and optical studies mapped epicardial surfaces; hence one may assume that activation wavefronts are still present intramurally or in the Purkinje fibers during isoelectric window, and the focal pattern of the epicardial activation for the first post-shock beat represents epicardial breakthrough from the deeper layer of the ventricle. A simulation study [36] demonstrated that, during the isoelectric window, an activation wavefronts induced by VEPs within the deep ventricular wall propagated fully intramurally through an excitable tunnel, until it emerged onto the epicardium, forming the focal pattern of the first post-shock epicardial activation. However, three-dimensional whole-heart mapping studies with plunge electrodes showed no evidence of wavefront propagation or reentry during the isoelectric window, and a new activation wavefront emerged focally following the isoelectric window [33, 35]. It is possible that plunge needle electrodes could not detect slowly propagated graded responses or were too few recordings to demonstrate activations in the intramural regions or the Purkinje system. However, we found that the isoelectric window followed by a focal discharge was still observed using an optical mapping technique in rabbit hearts with endocardial cryoablation in which only a thin layer (≈0.5-mm) of surviving epicardial tissues [37]. Therefore, the experimental evidence available supports the idea that the near DFT shocks eliminate all VF activations. A new wavefront of the focal mechanism rather than reentry is re-induced by the defibrillation shock in failed defibrillation.
Role of Intracellular Ca2+ in the Generation of Postshock Focal Activation
We simultaneously mapped epicardial membrane voltage (Vm) and Cai during attempted defibrillation at a near DFT shock strength in Langendorff-perfused rabbit ventricles [37]. We found that the heterogeneous post-shock distribution of Cai plays a key role in defibrillation failure. A focal activation following the isoelectric window in unsuccessful defibrillation always originates from the regions of low Cai surrounded by elevated Cai (i.e. Cai sinkholes) (Fig. 13.3a). No Cai sinkholes are present after type A successful defibrillation. We also determined the relationship between the Cai sinkholes and vulnerability to VF, since re-induction of VF by a defibrillation shock itself is responsible for defibrillation failure. The Cai sinkholes were also present after a shock on T-wave that induced VF (Fig. 13.3b) [37, 38]. What is the mechanism by which the first post-shock activation consistently arises from the Cai sinkhole? The first post-shock activation following the isoelectric window is focal, suggesting triggered activity as a potential mechanism. In some episodes of unsuccessful defibrillation, an earlier rise in Cai than Vm are observed at the early site for the post-shock focal activation. The ratio map shows that a rise in Cai precedes a rise in Vm at the Cai sinkhole site (i.e. Cai prefluorescence, arrow in Fig. 13.4). It is possible that the Cai prefluorescence might be attributable to spontaneous (voltage-independent) Ca2+ release from the sarcoplasmic reticulum (SR) that induces triggered activity by the reverse excitation-contraction coupling [39]. The low Cai in the sinkhole reflects more complete SR Ca2+ uptake. With the SR more Ca2+ loaded and more fully recovered, it may be more susceptible to spontaneous Ca2+ release from the SR, compared with regions with persistently high Cai. Consistent with this hypothesis, suppression of SR function by ryanodine and thapsigargin significantly reduced the upper limit of vulnerability and DFT [37]. Alternatively, EAD might be another explanation for the triggered activity. At the Cai sinkhole site, the Cai continues to decline, which promotes the recovery of ICa,L from inactivation. Reactivation of ICa,L increases both Cai and Vm, leading to EAD and triggered activity. Another possible mechanism is the electrotonic interaction with surrounding cells. In the Cai sinkholes, APD is shorter than in the surrounding sites, possibly because the reduced Cai level in the sinkholes may shorten the APD by the bidirectional coupling between Cai and Vm [40, 41]. Electrotonic currents from the surrounding depolarized regions might assist impulse formation in the sinkhole region in the similar way to phase 2 reentry or reflection [42].
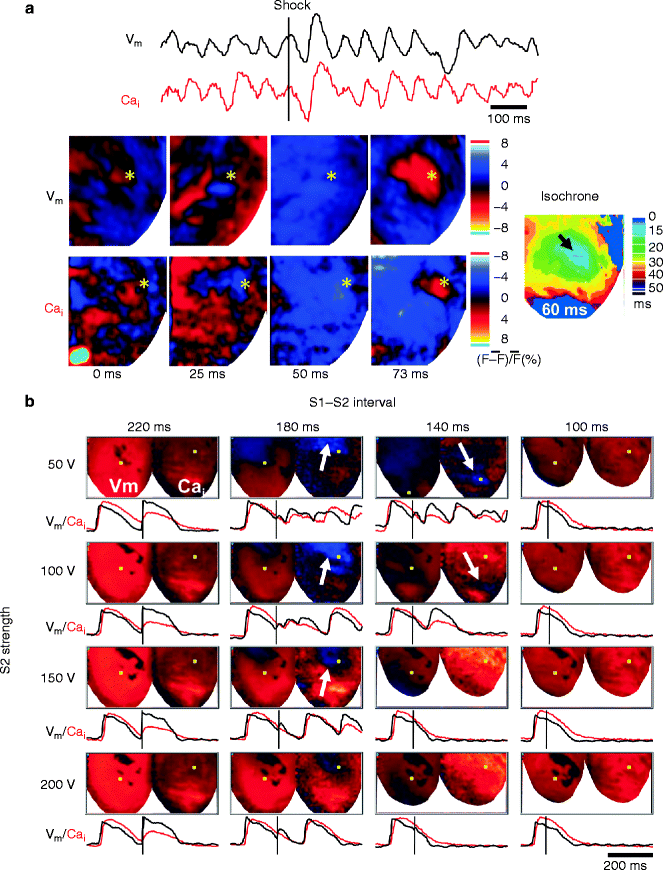
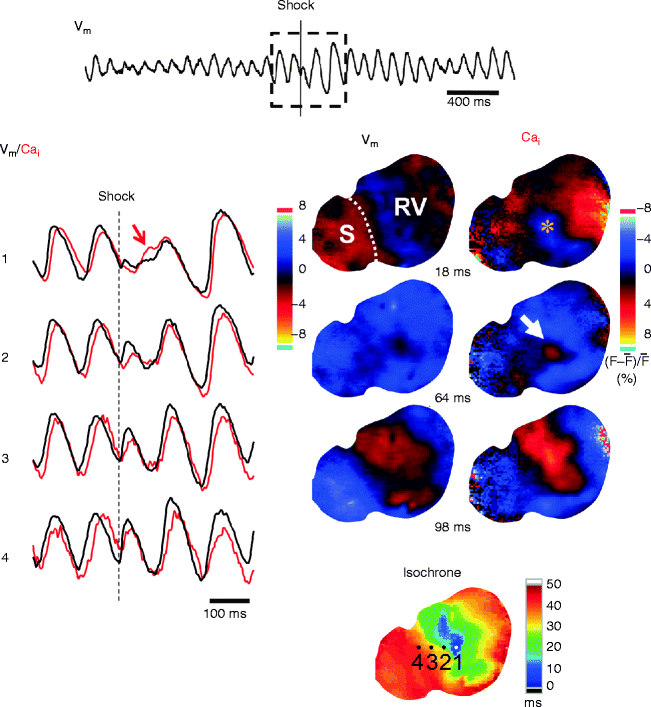
Figure 13–3.
Cai sinkhole in unsuccessful defibrillation (a) and when shock was given during the vulnerable period (b) in a Langendorff-perfused rabbit ventricle. (a) Epicardial Vm (black line) and Cai (red line) optical signals from the site marked by asterisk in snapshots of ratio maps. Electric shock at near DFT strength was applied at time zero (vertical line). After the shock, the Cai map showed an area with continued decline of Cai, resulting in a blue region on the Cai map (marked by asterisks in the frame 25–50 ms). This blue region had a lower Cai level than the surrounding tissues (Cai sinkhole). Thereafter, the entire Vm map showed repolarization coded with blue (frame 50 ms), consistent with the isoelectric window. Note that a focal discharge started at the Cai sinkhole site. (b) Shock on T wave with different S1–S2 coupling intervals and S2 shock strength. Note that all episodes associated with induction of VF or repetitive responses had the Cai sinkhole (white arrow) after the shock (From Hwang et al. [37] with permission)
Figure 13–4.
Cai prefluorescence at the Cai sinkhole after unsuccessful defibrillation in the endocardial surface of a Langendorff-perfused rabbit ventricle. Local Vm (black lines) and Cai (red lines) optical signals were obtained at sites indicated in the isochronal map for the post-shock first activation. The Cai sinkhole (asterisk) was observed (frame 18 ms) after shock at a near DFT strength. Note a rise in Cai (white arrow, frame 64 ms) while Vm map was still in a quiescent period (Cai prefluorescence), followed by a focal post-shock activation. At this site (site 1), a local Cai elevation (red region in frame 64 ms) preceded a rise in Vm. S interventricular septum, RV right ventricle
Biphasic Defibrillation
The heterogeneity of Cai as well as that of Vm immediately after shock seems important in the mechanisms of defibrillation and vulnerability, because it facilitates the formation of Cai sinkholes. It is known that biphasic waveform shock is more effective than monophasic waveform shock in achieving successful defibrillation. We demonstrated that the greater efficacy of biphasic waveform is directly related to less heterogeneous post-shock Cai distribution, thus reducing the probability of Cai sinkhole formation and VF re-initiation [43]. The electric field created by defibrillation shock alters the Vm but it also significantly changes the Cai [44, 45]. Transient hyperpolarization at virtual anodes is expected to increase the driving force for Ca2+ entry through ICa,L, potentiating SR Ca2+ release, while transient depolarization at virtual cathodes is not expected to exert this effect [46]. It follows that a shock-induced APD prolongation is more accentuated at the virtual anode sites than at the virtual cathode sites, since the increased Cai prolonged APD by potentiating inward Na+-Ca2+ exchange current (I NCX), consistent with positive Cai-APD coupling [43, 47]. During VF, the effects of VEPs on Cai are variable, because effects of shocks on Cai depend on the timing of shock application relative to the local repolarization state [43, 45]. Thus, the sites of the Cai sinkhole are not predictable by distribution of the VEPs, but the heterogeneous effects of VEPs on Cai would promote the Cai sinkhole formation. This may account for the more reduction in the Cai sinkholes by the biphasic waveform shocks which have a neutralizing effect on the VEPs.
Intracellular Ca2+ Dynamics and Immediate Recurrences of VF After Successful Defibrillation
Defibrillation failure can be viewed as an immediate recurrence of VF after the shock halts all VF activations. This type of VF recurrence generally occurs 50–100 ms after the shocks [23, 24, 37]. However, as documented by Zoll et al. in their first paper on electrical defibrillation [1], VF can also recur seconds after an electrical countershock that was initially successful. More recent studies show that spontaneous recurrences of VF can occur 1 s to 2 min after defibrillation for long-duration VF with resultant ischemia [48]. Also, defibrillation during global ischemia is often followed by immediate VF recurrences (<10 s after shock) [49]. Importantly, when the VF recurrences occur during ECG saturation caused by the strong electric shock, it masquerades as defibrillation failure. This implies that a shock-resistant VF, which is often observed in the out-of-hospital sudden cardiac arrest [50], may include this type of the pseudo-defibrillation failure. Furthermore, it has been reported that recurrent VFs in patients with a normal structural heart frequently arise from the distal Purkinje system [51]. In canine electrical mapping studies, Purkinje fibers are highly active and the earliest post-shock activation always originates from the Purkinje fibers after long-duration VF [48]. We recently found that triggered activity due to delayed afterdepolarization (DAD) is the mechanism of post-shock ventricular arrhythmias originating from the Purkinje fibers which have a higher Cai-Vm coupling gain due to a smaller IK1 [52].
Upregulation of I KAS in Heart Failure and Electrical Storm
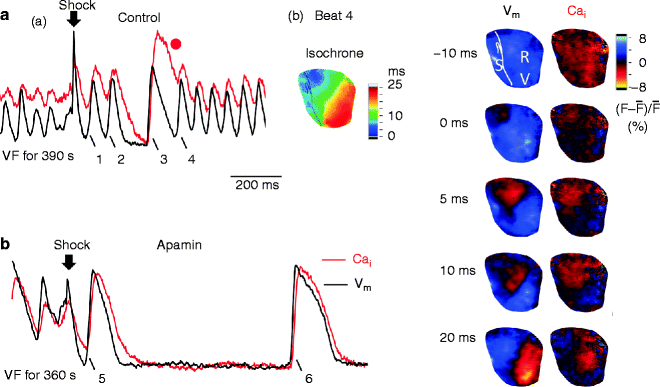
Recurrent spontaneous VFs after successful defibrillation are observed frequently in patients with heart failure [53]. We recently developed a rabbit model with pacing-induced heart failure which shows recurrent episodes of spontaneous VF [54, 55]. Simultaneous mapping of Vm and Cai in the failing hearts identified that marked APD shortening and disproportionally persistent Cai transient after successful defibrillation induce afterdepolarizations during late phase 3 of the action potential possibly by promoting the forward mode of I NCX (i.e. CCTF or late phase 3 EAD) [9–11], which causes triggered activity and re-initiation of VF. The importance of intracellular Ca2+ handling in electrical storm is also supported by the study by Tsuji et al. [56]. In that study, the authors found abnormal protein phosphorylation may affect the Ca2+ handling and contribute to the electrical storm in rabbits with complete heart block. In addition to abnormal Cai accumulation, shortened APD is also a major factor that promotes electrical storm. We discovered that in heart failure, there is a significant upregulation of apamin-sensitive potassium current (I KAS) conducted through the SK channels. Because of the Cai accumulation during VF, this channel is activated to conduct I KAS and shorten the APD. Inhibiting I KAS by apamin can suppress recurrent VF in our model (Fig. 13.5). Patch clamp studies confirmed the upregulation of I KAS in failing rabbit ventricles (Fig. 13.6). Our recent studies documented that SK channel is upregulated in failing human ventricles [57]. Further studies of I KAS may contribute to the better understanding of the mechanisms by which VF begets VF.