Fig. 17.1
Metabolic changes in advanced heart failure. Cardiovascular diseases such as hypertension, coronary artery disease, cardiomyopathy, and cardiac arrhythmia can lead to heart failure. In end-stage heart failure, typical metabolic changes are decreased fatty acid utilization, glucose utilization, and overall oxidative metabolism, resulting in impaired energetics. Effects of heart failure on systemic metabolism include insulin resistance, increased catabolism, and altered adipokine secretion, which contribute to the progression of heart failure and further impair myocardial energy metabolism. PCr:ATP is phosphocreatine:adenosine triphosphate
The derangement of cardiac energy metabolism in heart failure occurs at three stages – energy substrate utilization (uptake and oxidation), oxidative phosphorylation in mitochondria, and high-energy phosphate (ATP, phosphocreatine) metabolism, as was evidenced in the rodent and human heart [21]. Previous studies have generally shown that while fatty acid utilization is not substantially altered in early stages of heart failure [27, 28], it drops substantially in advanced heart failure [29] (Fig. 17.1). Glucose utilization has been reported to increase in early stages of heart failure [30] and decrease along with the development of insulin resistance in advanced heart failure [21, 31–33] (Fig. 17.1). Since the systemic metabolic milieu is drastically altered in heart failure with commonly increased circulating fatty acids, glucose, and insulin, and catabolic over activity, these results need to be viewed with caution as it is difficult to distinguish between the inherent impairment of substrate metabolism in the myocardium and metabolic adaptations due to altered substrate availability [21, 25]. Impaired structure and function of mitochondria are also commonly observed in failing hearts [34, 35], leading to reduced oxidative phosphorylation, oxygen consumption, and ATP/phosphocreatine production.
In addition, the creatine kinase system is substantially altered in heart failure [21, 36, 37], which causes a drastic decline in ATP transfer and ATP starvation of myofibrils. Interestingly, ATP concentrations are sustained in a normal range in earlier stages of heart failure and only decline by approximately 30–40 % in advanced heart failure [21, 38–40]. However, the decline in intracellular creatine and phosphocreatine due to impaired creatine transporter function precedes and is greater than the decline in ATP concentrations (30–70 %), representing an early sign of deranged cardiac energetics in heart failure [36, 39, 41, 42]. Hence, phosphocreatine-to-ATP ratios are reduced in heart failure and correlate with heart failure classes according to the New York Heart Association [43] (Fig. 17.1). These changes in high-energy phosphate metabolism drastically limit the energetic reserve of the myocardium. For example, when failing hearts are challenged with high workload (e.g., by stimulation with catecholamines), free ADP increases to concentrations that are double compared to those in the healthy heart [44], thereby reducing the contractile reserve [21]. Given that impaired energy metabolism critically contributes to heart failure development and/or progression, modulators that improve the energetic reserve of the heart may become attractive options for the treatment of heart failure.
At present, there is no approved therapy available that specifically targets energy metabolism in heart failure [25]. Experimental metabolic therapies that are aimed at improving metabolic balance and efficiency in the myocardium generally decrease fatty acid utilization and increase glucose oxidation [25]. For example, the piperazine derivative, trimetazidine, is an experimental drug that selectively inhibits mitochondrial long-chain 3-ketoacyl-CoA thiolase, thereby decreasing fatty acid oxidation and increasing glucose utilization via secondary activation of pyruvate dehydrogenase [45]. Trimetazidine restores coupling between glycolysis and glucose oxidation and leads to ATP production with less demand for oxygen [45]. While the results from clinical trials are promising [45–47], more clinical studies are required to demonstrate the efficacy of this drug in ameliorating heart failure and angina (Chap. 22).
Heart failure not only affects myocardial energy metabolism but whole body metabolism via endocrine communication of the heart with other organs [25, 48, 49]. For example, systemic insulin resistance is a characteristic feature of heart failure [48]. It develops in response to neurohormonal stimuli (catecholamines), inflammatory cytokine release, oxidative stress, and tissue hypoperfusion as a consequence of heart failure [25, 50]. Moreover, insulin resistance appears to predict severity of heart failure and reduced survival [25]. In addition, resistance to the anabolic hormone insulin, among other factors, leads to an overall catabolic environment that contributes to muscle cachexia in heart failure patients [25, 49]. Overstimulation of adipose tissue lipolysis via increased catecholamines, inflammatory cytokines, natriuretic peptides, and pressure overload also leads to a rise in circulating fatty acids and changes in adipokine secretion and thereby contributes to the systemic and cardiac metabolic imbalance in heart failure [25, 50, 51]. Therefore, therapies that target not only cardiac metabolism but whole body metabolism, for those individuals in heart failure, could hold promise in treating this debilitating disease (Chaps. 3 and 18).
17.3 Metabolic Remodeling in Myocardial Hypertrophy
Cardiac hypertrophy is an initially adaptive response to cellular stress leading to cardiomyocyte enlargement, increased protein synthesis, re-induction of the so-called fetal gene program, and heightened sarcomeric organization [52, 53]. Chronically, cardiac hypertrophy can become maladaptive and trigger heart failure and malignant arrhythmia due to perturbations of cellular calcium homeostasis and ionic currents [52, 53]. Significant morphological changes following long-term cardiac hypertrophy include increased rates of programmed cell death, fibrosis, and cardiac chamber dilatation [52, 53]. Common stressors that lead to hypertrophic remodeling in cardiomyocytes are pressure or volume overload, mutations of sarcomeric or other proteins, and loss of contractile mass from prior infarction [52, 53]. The following section describes metabolic changes following pressure overload hypertrophy as this type of hypertrophy is increasingly common due to the increasing prevalence of hypertension (Chap. 16).
One of the metabolic hallmarks of pressure overload-induced cardiac hypertrophy is that cardiac energy metabolism reverts to a fetal-like profile, which is due to a decrease in fatty acid oxidation and increased reliance on carbohydrates for ATP production with an overall decrease in oxidative metabolism [54, 55] (Fig. 17.2). This substrate shift has been suggested to contribute to the progression of cardiac hypertrophy to overt heart failure [56], although it still remains elusive to what extent metabolic remodeling influences the development and progression of pressure overload-induced cardiac hypertrophy. The reduction in fatty acid oxidation is attributed to a decrease in the expression of genes involved in β-oxidation and oxidative phosphorylation [55]. A number of studies using animal models have shown that this is due to the downregulation of the transcriptional master regulators PPARα and PGC1 [55, 57–60] (Fig. 17.2). In addition, a reduction in membrane-bound fatty acid transporters and carnitine has also been observed in the hypertrophic heart [61–64]. The resulting energy insufficiency leads to the activation of the energy-sensing kinase, AMPK, which contributes to an increase in glucose uptake and glycolysis by promoting translocation of glucose transporters to the sarcolemma and stimulating the glycolytic enzyme, phosphofructokinase 2 [55, 65–67] (Fig. 17.2). Interestingly, the increase in glucose uptake is insulin-independent, and changes in glucose metabolism in the hypertrophic heart are not accompanied by marked changes in proteins involved in glucose transport or glycolysis [33, 55, 68]. In contrast to the increased glycolysis, many studies have reported that glucose oxidation is either unchanged or decreased in the hypertrophied heart, suggesting that glucose oxidation and glycolysis are uncoupled in cardiac hypertrophy [55, 69–71] (Fig. 17.2). As a result of this uncoupling in glucose metabolism, lactate dehydrogenase, which converts pyruvate into lactate, is activated to process the excess pyruvate. Consequently, increased secretion of lactate from the hypertrophied myocardium has been reported [55, 72, 73].
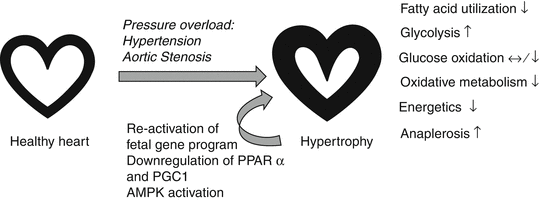
Fig. 17.2
Metabolic changes in pressure overload-induced cardiac hypertrophy. Pressure overload-induced hypertrophy, triggered by hypertension or aortic stenosis, is associated with a decrease in fatty acid oxidation and overall oxidative metabolism and energetics, while anaerobic glucose metabolism (glycolysis) is increased. Glycolysis is uncoupled from glucose oxidation, and excess pyruvate is shuttled towards alternative pathways, such as anaplerosis. The decrease in fatty acid utilization is mostly due to the reactivation of the fetal gene program and downregulation of transcriptional regulators of fatty acid oxidation and mitochondrial biogenesis and function. AMPK activation has been suggested to underlie the increase in glucose uptake and glycolysis in cardiac hypertrophy
The excess pyruvate can also be shuttled towards anaplerosis, which refers to metabolic processes that replenish tricarboxylic acid (TCA) cycle intermediates that are removed from the TCA cycle for biosynthetic pathways to produce glucose, fatty acids, and amino acids [74]. In this process, pyruvate is converted to oxaloacetate and malate through its carboxylation via pyruvate carboxylase and malic enzyme, respectively [74]. Consistent with this notion, an 80–90 % increase in anaplerotic flux has been reported in the hypertrophied heart [75, 76] (Fig. 17.2). Although pyruvate can replenish TCA cycle substrates via anaplerosis, it is an energetically costly process as it reduces the efficiency of ATP production from pyruvate [55]. Changes in other glucose metabolism pathways were also observed in the hypertrophied heart, including pentose-phosphate pathway [77–79] and hexosamine pathway [80, 81]. To date, it remains unclear whether and to what extent these “alternative” glucose metabolism pathways contribute to the pathophysiology of cardiac hypertrophy.
The increase in the reliance on glucose utilization in the hypertrophic heart appears to be an adaptive process, at least in earlier stages of hypertrophic remodeling. This notion is inferred from studies with mutant mice where the expression of glucose transporters has been altered. For example, mice overexpressing the insulin-independent glucose transporter Glut1, specifically in the heart, exhibit increased glucose uptake and glycolysis that is partially uncoupled from glucose oxidation, as well as decreased fatty acid oxidation [82]. Importantly, these mice were protected from pressure overload-induced cardiac dysfunction [82]. In contrast, mice with cardiac deficiency of the insulin-sensitive glucose transporter Glut4 exhibit reduced contractile function and cardiomyocyte hypertrophy [83]. Prevention of metabolic remodeling in pressure overload hypertrophy via cardiac-specific deletion of acetyl-CoA carboxylase 2, which produces the fatty acid oxidation inhibitor malonyl-CoA, attenuated cardiac hypertrophy, protected against fibrosis, and improved cardiac function [84]. These findings suggest that metabolic remodeling contributes to the pathophysiology of cardiac hypertrophy.
Interestingly, cardiac-specific overexpression of PGC1α and the associated increase in mitochondrial size did not protect from cardiac remodeling induced by transverse aortic constriction [85]. Instead, it led to a greater impairment in contractile function and increase in left ventricular chamber dimension [85]. These data suggest that attempts to stimulate master regulators of mitochondrial biogenesis and size in cardiac hypertrophy to improve oxidative metabolism may in fact worsen outcomes following pressure overload-induced hypertrophy. Dietary and pharmacological strategies have also been pursued to ameliorate outcomes in pressure overload-induced hypertrophy [54, 86, 87]. Interestingly, feeding rats with a diet low in carbohydrates and high in fat attenuated cardiac hypertrophy and remodeling [86]. In contrast, activation of fatty acid metabolism via PPARα agonist (WY-14643) treatment augmented pressure overload-induced contractile dysfunction, despite the prevention of substrate switching [87]. Since these interventions drastically influence whole body metabolism in addition to cardiac metabolism, their direct effects on metabolism and function in the hypertrophic heart remain unclear.
17.4 Metabolic (Mal)adaptation of the Heart in Obesity
Obesity, defined as excess accumulation of body fat, and associated type 2 diabetes mellitus, is a significant risk factor for the development of heart failure [88, 89]. Although the onset of heart failure in obesity is likely multifactorial, obesity-associated cardiomyopathy appears to be a major initiating factor and contributes to the increased morbidity and mortality among obese individuals [88]. This is exemplified by the finding that even after correcting for hypertension and other common obesity-related risk factors, the presence of obesity still approximately doubles the risk of developing heart failure [90]. Chronic obesity commonly leads to systemic metabolic perturbances including insulin resistance and type 2 diabetes with concomitant hyperglycemia and hyperlipidemia. It has been hypothesized that this oversupply of energy substrates to the heart initially leads to adaptive changes and ultimately precipitates contractile dysfunction [90]. Specifically, the increased availability of fatty acids resulting in augmented fatty acid uptake, in conjunction with inadequate activation of fatty acid oxidation, gives rise to excess accumulation of toxic lipid metabolites in the myocardium and a general increase in cardiac fat content [88, 90]. High fatty acid oxidation rates in obesity, as have been observed in both animal models and humans, inhibit cardiac glucose utilization via substrate competition, hence contributing to decreases in glycolysis and glucose oxidation as well as insulin resistance [19, 91] (Fig. 17.3). These metabolic changes also lead to a decrease in mechanical efficiency [19, 91]. It has been suggested that the metabolic remodeling of the myocardium observed in obesity not only precedes but contributes to overt functional and structural changes of the heart in obesity [90, 92].
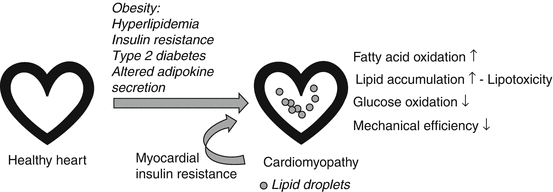
Fig. 17.3
Metabolic changes in obesity-associated cardiomyopathy. Obesity and obesity-associated systemic changes in energy metabolism (hyperlipidemia, insulin resistance, type 2 diabetes) and adipokine secretion lead to increased cardiac fatty acid oxidation, lipid accumulation, and lipotoxicity, which are paralleled by reduced glucose oxidation and mechanical efficiency. Myocardial insulin resistance further promotes metabolic dysregulation in the heart
Increased accumulation of toxic lipid metabolites, a process that is also termed as “lipotoxicity,” has also been suggested to contribute to cardiac dysfunction during obesity [93–95] (Fig. 17.3). Examples for toxic lipid metabolites are long-chain acyl-CoAs, ceramides, diacylglycerols, and acylcarnitines [96]. Lipotoxicity may lead to cardiac dysfunction by means of activating apoptosis, impairing insulin signaling, promoting endoplasmic reticulum stress, activating protein kinase C and mitogen-activated protein kinase, as well as modulating PPAR signaling [96]. Studies using nonobese transgenic mice and obese-diabetic rat models show that accumulation of lipids in cardiomyocytes corresponds with a decrease in systolic and diastolic function and cardiac hypertrophy [4, 19, 93, 97–101]. However, the individual contribution of lipid subspecies to cardiac pathophysiology is unclear. Mechanisms for the lipotoxicity-induced insulin resistance in the heart have been suggested to involve diacylglycerol-mediated activation of protein kinase C, resulting in increased serine phosphorylation of insulin receptor substrate 1 and decreased activation of downstream insulin signaling mediators, such as phosphatidylinositol 3-kinase and Akt [19]. The implication of increased TAG accumulation in the obese heart is less understood, but it may impair cardiac function by fueling the production of “toxic” lipid species [1]. Myocardial TAG content positively correlates with body mass index, suggesting that cardiac TAG deposition increases gradually with increased adiposity [102]. Elevated myocardial TAG content was also observed in individuals with impaired glucose tolerance and type 2 diabetes [1, 103, 104]. Moreover, the increase in cardiac TAG accumulation preceded the development of overt cardiac dysfunction, suggesting a causal relationship between elevated TAG levels in the heart and obesity/type 2 diabetes-associated myocardial dysfunction [1, 103].
To date, drugs used to treat metabolic disturbances in obesity are mainly aimed at lowering circulating lipid levels and ameliorating insulin resistance. For example, there are two classes of PPAR agonists used to achieve this effect. These are ligands for PPARα and PPARγ, respectively. Both PPARα and PPARγ agonists lower circulating lipid levels either by increasing fat storage in adipocytes or increasing fatty acid oxidation in the muscle and liver [19]. Despite the beneficial systemic effect of PPARα and PPARγ agonists, their direct effect on the heart may not always be desirable. For example, cardiac-specific overexpression of PPARα induced a cardiac phenotype similar to diabetic cardiomyopathy [105]. Myocardial fatty acid oxidation rates were increased in these transgenic mice, while glucose uptake and oxidation were decreased, concomitant with the development of pathological cardiac hypertrophy, increased lipid accumulation, and cardiac dysfunction [105]. Similarly, cardiomyocyte-specific overexpression of PPARγ led to cardiac dysfunction in mice that was associated with increased myocardial lipid accumulation and expression of enzymes involved in fatty acid utilization [106]. In humans, PPARγ agonist treatment is also associated with peripheral edema and heart failure [106, 107]. These findings suggest that PPAR agonist treatment in obesity may have adverse effects on the heart by increasing cardiac lipid deposition. Interestingly, recent studies showed that inhibition of mitochondrial β-oxidation with trimetazidine, which is widely used for the treatment of angina, can improve obesity-related cardiac dysfunction [108]. Trimetazidine not only improves contractile efficiency in obese humans but protects against obesity-induced systolic and diastolic dysfunction in mice without altering insulin sensitivity or exacerbating obesity-induced insulin resistance [108]. This suggests that trimetazidine, by improving the metabolic balance in the heart, may be a viable therapy for the treatment of obesity-related cardiomyopathy.
Metabolic disturbances in adipose tissue initiate obesity-associated morbidity and cardiovascular disease [109, 110]. Besides fueling the systemic and cardiac lipid oversupply in obesity, the hypertrophic obese adipose tissue also changes its secretory profile of hormones and cytokines, so-called adipokines, which promotes not only insulin resistance and inflammation but likely has a direct effect on cardiac energy metabolism [19, 90, 111] (Fig. 17.3). However, more studies are required to better understand the direct effects of changes in circulating adipokines such as adiponectin, leptin, resistin, and retinol-binding protein 4 on cardiac metabolism and function during obesity in both animal models and humans.
17.5 Concluding Remarks
Cardiac energy metabolism is tightly regulated to meet the high energy demands of myocardial contraction and to adapt to short-term fluctuations in energy substrate supply and workload. When challenged by chronic stressors, including pressure overload and obesity, myocardial energy metabolism can initially adapt to counter-regulate but is eventually locked into a dysregulated and inflexible state with a major shift in substrate utilization and/or cardiac energetics and efficiency. Many studies have shown that this impairment in cardiac energy metabolism can promote the development of heart failure with a significant drop in the heart’s energetics and premature death. Since there is evidence that maladaptive changes in cardiac energy metabolism contribute at least in part to multiple forms of heart disease, drugs that aim to restore a balanced substrate utilization in the heart have the potential to become attractive options to treat these diseases and prevent their progression to overt heart failure.
References
1.
Kienesberger PC, Pulinilkunnil T, Nagendran J, Dyck JR. Myocardial triacylglycerol metabolism. J Mol Cell Cardiol. 2013;55:101–10. PubMed PMID: 22789525, Epub 2012/07/14. eng.PubMed
2.
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90(1):207–58. PubMed PMID: 20086077, Epub 2010/01/21. eng.PubMed
3.
Pulinilkunnil T, Abrahani A, Varghese J, Chan N, Tang I, Ghosh S, et al. Evidence for rapid “metabolic switching” through lipoprotein lipase occupation of endothelial-binding sites. J Mol Cell Cardiol. 2003;35(9):1093–103. PubMed PMID: 12967632, Epub 2003/09/12. eng.PubMed
4.
Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y, et al. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest. 2003;111(3):419–26. PubMed PMID: 12569168, Pubmed Central PMCID: 151861, Epub 2003/02/06. eng.PubMedCentralPubMed
5.
Heather LC, Pates KM, Atherton HJ, Cole MA, Ball DR, Evans RD, et al. Differential translocation of the fatty acid transporter, FAT/CD36, and the glucose transporter, GLUT4, coordinates changes in cardiac substrate metabolism during ischemia and reperfusion. Circ Heart Fail. 2013;6(5):1058–66. PubMed PMID: 23940308.PubMed
6.
Harmon CM, Luce P, Beth AH, Abumrad NA. Labeling of adipocyte membranes by sulfo-N-succinimidyl derivatives of long-chain fatty acids: inhibition of fatty acid transport. J Membr Biol. 1991;121(3):261–8. PubMed PMID: 1865490.PubMed
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


