Fig. 5.1
Aldosterone exerts effects in the kidney, the heart, the vasculature and on the metabolism. It binds to the mineralocorticoid receptor in various target cells to induce different effect mechanisms
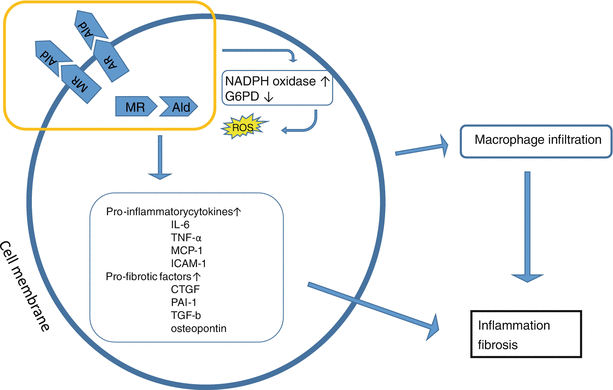
Fig. 5.2
Stimulation of cytosolic and membrane MRs by aldosterone in combination with stimulation of aldosterone receptors in the cell membrane lead to pro-inflammatory and pro-fibrotic effects [2]. In the mitochondria NADPH oxidase activity is increased, while G6DP activity is reduced, stimulating ROS generation [3]. Furthermore aldosterone upregulates pro-inflammatory and pro-fibrotic cytokines, and induces macrophage infiltration [4]. Ultimately these changes lead to tissue inflammation and fibrosis. MR mineralocorticoid receptor, Ald aldosterone, AR aldosterone receptor, G6PD glucose-6-phosphate dehydrogenase, ROS reactive oxygen species; IL-6 interleukin; TNF-α tumor necrosis factor, MCP-1 monocyte chemoattractant protein, ICAM-1 intercellular adhesion molecule, CTGF connective tissue growth factor, PAI-1 plasminogen activator inhibitor, TGF-b transforming growth factor
Aldosterone is pathophysiologically involved in kidney damage through multiple mechanisms in addition to the effects of elevated systemic blood pressure [4, 5] (Fig. 5.2). Firstly, glomerular damage is induced by the increased production of reactive oxygen species (ROS) by mitochondria [6, 7]. Furthermore, aldosterone increases expression of the pro-inflammatory serine/threonine-protein kinase and activates NFkB, which ultimately lead to mesangial inflammation, fibrosis, and glomerular injury [8]. Also macrophage infiltration into the renal cortex is stimulated by aldosterone, which promotes inflammation [9]. Lastly increased local renal aldosterone production has been shown to induce apoptosis in podocytes, through mechanisms which are not completely elucidated [10]. These local effects were larger in rats with diabetes mellitus, which was associated with increased MR and aldosterone levels. All the mechanisms listed above may contribute to renal damage and consequent deterioration of kidney function. Indeed, an increased proteinuria has been found in patients with excess aldosteronemia, independent of blood pressure [4, 5]. As proteinuria is a bad prognostic sign for CKD, this adds to the rationale for aldosterone inhibition as a new promising approach in halting disease progression.
The heart was the first extra-renal organ where mineralocorticoid receptors were found. Here aldosterone has been shown to promote electrophysiological remodeling, leading to atrial fibrillation and ventricular arrhythmias [3]. Remodeling takes place through modulation of T-type, potassium L-type and ryanodine receptor calcium channel activity [11–14]. Furthermore through pro-inflammatory processes cardiac hypertrophy and cardiac fibrosis are propagated, resulting in a reduced cardiac function. The blood vessels are a target for aldosterone through MRs in the vascular smooth muscle cells and endothelial cells. Here too hyperaldosteronism can lead to tissue inflammation, remodeling and fibrosis. MRA leads to endothelial swelling and stiffness by promoting the insertion of the epithelial sodium channel (ENaC) into the cell membrane [15]. Due to a simultaneous decreased ability to form nitric oxide, vasodilatation is limited. There is evidence that this is a main pathophysiological event leading to hypertension caused by hyperaldosteronism, rather than excess fluid retention.
Excess aldosterone levels are associated with the cardiometabolic syndrome, which is characterized by insulin resistance, central obesity, dyslipidemia and hypertension [3, 16]. Aldosterone impairs pancreatic beta cell function by promoting inflammation and oxidative stress in the islets of Langerhans [17, 18]. Also aldosterone degrades insulin receptor substrate proteins, reducing insulin sensitivity and glucose-uptake [19]. Furthermore, aldosterone promotes the release of inflammatory cytokines from adipose tissues resulting in systemic inflammation and impaired glucose tolerance [18]. The MR is found in adipocytes and perivascular adipose tissue (PVAT). Adipocytes and PVAT can produce local levels of aldosterone which exert paracrine and autocrine effects, influencing not only adipose tissues but also the vasculature and thus promoting vascular remodeling. In line, aldosterone blockade has been shown to be particularly effective to reduce blood pressure in obesity–related hypertension [20].
The Interaction Between Salt Status and Aldosterone Effects
The activity of the RAAS depends on what is denoted to as the “salt status”, the RAAS being suppressed in state of sodium/fluid excess and being activated in a state of sodium depletion. Interestingly, an interaction between salt status and the pro-fibrotic, pro-inflammatory effects of aldosterone has been described, which is independent of blood pressure [21]. While in rats on a high or normal salt diet, high levels of aldosterone are associated with development of substantial end organ damage, high levels of aldosterone do not have such target organ effects during a low salt diet. Furthermore, the hypertrophic effects observed in high-salt, high-aldosterone rats can be completely reversed by the addition of MRA inhibitors, underlining the causal role of the mineralocorticoid receptor in the pathophysiological process. Salt excess has a role in sensitizing cardiovascular tissue for damage caused by an excess in aldosterone through mechanisms currently unknown. Interestingly new research shows that in CKD patients urinary salt excess is a significant predictor of urinary excretion of the mineralcorticoid metabolites tetrahydroaldosterone and tetrahydrocorticosterone, suggesting an alternative regulatory mechanism for aldosterone [22]. Whereas this novel insight may provide a missing link between CKD, high salt-status, and increased target organ damage, further research regarding this topic is warranted.
MRA Inhibition
MRA Inhibition in Heart Failure
After publication of the results from the large randomized controlled trials RALES and EPHESUS, MRA inhibition has been adopted as part of the standard treatment of chronic heart failure [23, 24]. These studies showed that MRA inhibition with spironolactone or eplerenone reduces morbidity and mortality in patients with severe chronic heart failure (NYHA functional class III and IV; Table 5.1). The EMPHASIS-HF study showed that eplerenone was also effective in patients with mild symptoms of heart failure (NYHA functional class II) [25]. This is supported by a meta-review published in 2009 which showed a reduction in all-cause mortality and an increase in ejection fraction in patients with left ventricular failure on spironolactone therapy [30]. However, in a recent observational study spironolactone was shown not to have a significant effect on risk of hospitalization and death, suggesting that the benefits of MRA inhibition that has been found in randomized trials may not be true in clinical practice [26]. However, in this study outcome was not adjusted for baseline disease severity and spironolactone use may have been more often applied in patients with a worse clinical condition. Currently the ACCF/AHA recommends MRA inhibition therapy in patients with NYHA class II to IV heart failure and a left ventricular ejection fraction of ≤35 %, unless contraindicated [31].
Table 5.1
Long term effects of MRA inhibition in several types of disease
Author | Study type | Studied population | No of patients | Baseline medication | MRA | Follow up | Endpoints | Results | Hyperkalemia |
|---|---|---|---|---|---|---|---|---|---|
Heart failure | |||||||||
Pitt et al. [23] | Randomized controlled trial | NYHA III and IV heart failure | 1,663 | ACEi, loop diuretic | Spironolactone 25 mg daily | 24 months | Death from any cause | RR of death, 0.70; 95 % CI 0.60–0.82, P < 0.001; RR of hospitalization, 0.65; 95 % CI 0.54–0.77, P < 0.001 | Minimal incidence of serious hyperkalemia |
Pitt et al. [24] | Randomized, double-blind, placebo-controlled trial | Acute myocardial infarction complicated by LV dysfunction and heart failure | 6,632 | Optimal treatment | Eplerenone 25–50 mg daily | 16 months | Death from any cause | RR of death, 0.85; 95 % CI 0.75–0.96, P = 0.008 | Serious hyperkalemia, 5.5 vs 3.9 % in the placebo group (P = 0.002) |
Zannad et al. [25] | Randomized, double-blind placebo-controlled, parallel-group trial | NYHA II heart failure | 2,737 | ACEi, ARB, or both, and a beta-blocker | Eplerenone up to 50 mg daily | 21 months | Death from CV causes or a first hospitalization for HF | HR of death from CV cause or first hospitalization for HF, 0.63; 95 % CI 0.54–0.74, P < 0.001 | Serious hyperkalemia, 11.8 vs 7.2 % in the placebo group P < 0.001 |
Lee et al. [26] | Prospective cohort study | Newly diagnosed heart failure, LVEF <40 % | 2,538 | ACEi, ARB, beta-blocker, loop diuretic and/or calcium channel blocker | Spironolactone | 36 months | Death from any cause, hospitalization | Adjusted HR of death, 0.93, 95 % CI 0.60–1.44; adjusted HR of hospitalization 0.91, 95 % CI 0.77–1.08 | Severe hyperkalemia 4.8 per 100 person-years vs 1.6 per 100 person-years with nonuse, p < 0.001 |
Chronic kidney disease | |||||||||
Navaneethan et al. [27] | Cochrane systematic review | CKD patients | 845 | ACEi or ARB | Spironolactone | 2–12 months | Proteinuria, GFR, BP | 24 h proteinuria, 7 studies, 372 patients; MD −0.80 g, 95 % CI −1.23 to −0.38. eGFR, 5 studies, 306 patients: MD −0.70 mL/min, 95 % CI −4.73 to 3.34. Systolic BP, 7 RCTs, 372 patients: MD −3.40 mmHg, 95 % CI −5.13 to −1.68. Diastolic BP, 6 studies, 336 patients: MD −1.79 mmHg, 95 % CI −2.99 to −0.59 | Hyperkalemia, 8 studies, 436 patients: RR 3.06, 95 % CI 1.26–7.41 |
Mavrakanas et al. [28] | Systematic review | Diabetic nephropathy | 404 | ACEi or ARB | Spironolactone 25 mg daily or eplerenone 50–100 mg daily | 2–12 months | Proteinuria, GFR, BP | Reduction of albuminuria by 23–67 %. GFR slightly decreased. Significant drop in BP | Increased incidence hyperkalemia |
Boesby et al. [29] | Open randomized cross-over trial | Non-diabetic chronic kidney disease | 40 | ACEi or ARB | Eplerenone 25–50 mg daily | 2 months | Albuminuria, creatinine clearance, BP | Urinary albumin excretion was 22 % lower, CI: 14,28, P < 0.001; creatinine clearance 5 % lower, CI: 2,8, P = 0.005; mean systolic BP 4 mmHg lower, CI: 2,6, P = 0.002; diastolic BP 2 mmHg lower, CI: 0,4, P = 0.02 | Potassium 0.1 mEq/L higher, CI: 0.1, 0.2, P < 0.001 |
The Role of MRA Inhibition in CKD Treatment
Current treatment of CKD aims to prevent progressive renal function loss and its associated cardiovascular complications. Treatment of hypertension and proteinuria is the cornerstone of renoprotection. Inhibition of the RAAS by either ACEi or ARB has been proven to be an effective treatment option in CKD, reducing proteinuria as well as the rate of renal function loss. However, additional treatment options are necessary as progression of kidney disease still occurs in many patients. Combined blockade of the RAAS at different levels has been tested in several combinations, mostly ACEi combined with ARB [32]. Whereas dual blockade, with either ACEi plus ARB, or ACEI plus a renin-inhibitor is associated with a better efficacy on short term (i.e. proteinuria), hard outcome studies show that dual RAAS blockade does not confer better reno- and cardio-protection, but to the contrary, is associated with worse outcome [33].
Another strategy to improve the efficacy of RAAS-blockade, is in manipulating the salt status: correction of the volume overload in patients on monotherapy RAAS-blockade, by either diuretics, a low sodium diet or their combination, considerably potentiates the efficacy of monotherapy RAAS-blockade [34, 35, 36]. Post-hoc analyses suggest that moderate dietary sodium restriction is also associated with better long-term outcome of RAAS-blockade [37], but prospective studies, so far, are unfortunately lacking. Observational data have shown a worse long term outcome in subjects that consume very low (<5 g/day) amounts of salt [38]. Whereas this might be related to underlying conditions associated with poor intake and malnutrition, it has also been pointed out that reactive hyperreninemic hyperaldosteronism might exert an adverse effect in such subjects [39] (Fig. 5.3). Of note, despite interference with the renin-angiotensin axis, a low sodium diet during ACEi or ARB is associated with secondary hyperreninemic aldosteronism that is particularly marked during the combination of diuretics and low sodium, demonstrating that the feedback loop between volume status and aldosterone is not disrupted by the current modes of inhibition of the RAAS by ACEi or ARB.
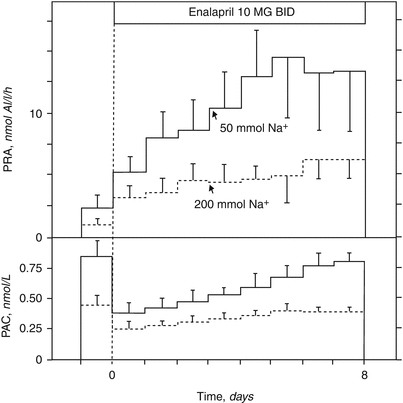
Fig. 5.3
Effects of enalapril on PRA and PAC (mean ± SEM) on liberal (broken lines) and low (continuous lines) sodium diet. PRA plasma renin activity, PAC plasma aldosterone concentration (Reprinted by permission from Macmillan Publishers Ltd: Navis et al. [39])
It has long been known that while under RAAS inhibition treatment with ACEi or ARB, aldosterone levels initially decrease, but can thereafter gradually increase to pretreatment, or even exceed pretreatment levels, a phenomenon that has been called “aldosterone breakthrough” or “aldosterone escape” [40] (Fig. 5.3). Whereas this so-called escape is usually associated with long term treatment, detailed assessment of the early effects of RAAS-blockade on aldosterone levels demonstrate that, actually, aldosterone displays an early partial return towards its baseline values even during the first week of treatment, possibly in response to the negative sodium balance induced by the RAAS-blockade, again demonstrating the preservation of aldosterone’s role in volume homeostasis, despite ACE-inhibition [41].
Keeping in mind the deleterious effects of aldosterone on the kidneys, MRA inhibition is a promising new therapeutic approach in CKD. Studies in experimental animals, in proteinuric models and in CKD patients support a renoprotective effect of MRA inhibition. As monotherapy RAAS-blockade is therapy of choice for CKD, in particular the added effects of MRA to ACEi or ARB are of clinical interest. These could be due to interference with classic effects of aldosterone (i.e. a diuretic effect of MRA) as well as due to interference with the non-classic effects of aldosterone. A Cochrane review dating from 2009 has shown that MRA antagonism in addition to an ACEi or ARB, can in small doses significantly reduce proteinuria while only slightly influencing renal function or blood pressure, with the adverse effect of an increased incidence of hyperkalemia (Table 5.1) [27]. Long term clinical endpoints such as cardiovascular events or renal function were not available in the included studies. A recent systematic review concerning the effect of MRA inhibition in addition to RAAS inhibition on diabetic nephropathy confirmed these effects on proteinuria, blood pressure, glomerular filtration rate and potassium levels, however data on long term cardiovascular and renal outcome were still not available [28]. In non-diabetic patients with CKD, long term effects of MRA inhibition in addition to RAAS inhibition are also unavailable, while short term effects are comparable to that in patients with diabetic nephropathy [29]. In patients with mild CKD, treatment with MRA inhibitors was shown to have a beneficial effect on left ventricular mass and arterial stiffness [42]. In summary, the short-term effects of spironolactone and eplerenone are beneficial for CKD patients and these agents are also safe, provided that potassium levels are being monitored. However, there is a lack of data concerning the long term clinical outcome of dual treatment with an ACE-inhibitor or ARB and a MRA inhibitor, as well as lack of data on the mechanism of the added effect of MRA to ACEi or ARB. The treatment can be considered in individual patients who otherwise have an unacceptable poor (renal) prognosis, given that these patients are extremely well instructed with regards to stopping the MRA inhibitor in circumstances of volume/sodium depletion. It is obvious that more research is needed to determine the definite role of MRA inhibition in CKD treatment, in particular whether the risks outweigh te benefits.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


