Pathophysiology and epidemiology
1. Poor understanding of the pathophysiology in AHFS
2. Heterogeneous patient population in terms of pathophysiology, etiology, and clinical presentation
3. Uncertain relationship between hemodynamics and neurohormonal changes and outcomes
4. Clinical course (particularly soon after discharge) of AHFS has not been well studied
The therapy (experimental drug or device)
5. Cardiac and noncardiac comorbid conditions influence the outcome and interaction with therapy
6. The transition from animal studies to clinical studies has occurred without comprehensive understanding of the mechanistic properties of the drug in specific patient subgroups. Not “knowing” the drug
7. Not having the correct dose
8. Possible variation of efficacy and safety with time given significant fluctuations in symptoms, hemodynamics, neurohormones, renal function, and myocardial injury during the course of hospitalization and postdischarge, the efficacy and/or safety of the drug may be dependent on the time of the intervention
9. The majority of drugs tested thus far reduce systemic BP, which may potentially decrease coronary perfusion, thereby contributing to myocardial and/or kidney injury
The protocol
10. Patient selection
11. Surrogate end points and clinical outcomes in phase II do not predict the results of a phase III trial
12. Choice of end points
13. Because most patients’ signs and symptoms improve with standard therapy, it is difficult to prove that novel therapies are producing further and substantial improvements
Study execution
14. Selection of incorrect patients and/or less than ideal follow-up
Table 31.2
Summary of failed phase III studies
Phase III studies | Primary end point(s) | Phase III primary end point reached? | Contributors to lack of success per Table 31.1 |
|---|---|---|---|
Tolvaptan | 4,6,7,8,11 | ||
EVEREST (n = 4,133) | Long term | No | |
1. All-cause mortality (superiority and non-inferiority) | |||
2. Cardiovascular death or hospitalization for HF (superiority only) | |||
Short-term | |||
Composite of global clinical status and body weight reduction | Yes (driven by body weight reduction) | ||
Tezosentan | 2,3,4,5,7,8,9,10,13 | ||
VERITAS (n = 1,448) | 1. Change in dyspnea (visual analog scale) over 24 h (in the individual trials) | No | |
2. Death or worsening HF at 7 days (in both trials combined) | |||
Levosimendan | 3, 5,6,7,9,11 | ||
REVIVE-2 (n = 600) | Composite of clinical signs/symptoms of HF and | Yes—excess hypotension and arrhythmia with trends towards early mortality with levosimendan | |
1. Patient reported moderately or markedly improve at 6 h, 24 h, and 5 days (and no criteria for worsening) | |||
2. Worsening (death, patient reported moderate or severe deterioration at any time point) or worsening symptoms at any time or persistent severe symptoms after 24 h requiring rescue therapy (ie intravenous diuretic, vasodilator or inotropic agents) | |||
3. Unchanged | |||
SURVIVE (n = 1,327) | All-cause mortality at 180 days | No | |
PROTECT (n = 2,033) | Trichotomous classification of patients | No | |
1. Success: Improvement in dyspnea (Likert scale-moderately or markedly better) at 24 and 48 h or day of discharge, and not meeting criteria for treatment failure | |||
2. Failure: death, HF readmission within 7 days, worsening HF (by physician assessment by day 7), or persistent renal impairment | |||
3. Unchanged: neither criteria for success or failure | |||
Nesiritide | 3,4,7,8,9,12,13 | ||
ASCEND-HF | 1. Composite of all-cause mortality + HF rehospitalization at 30 days | No | |
2. Dyspnea at 6 and 24 h |
The International Working Group on Acute Heart Failure Syndromes has conducted a series of meetings at the United States Food and Drug Administration over the past 5 years, and we believe that we are now better equipped to conduct future studies with improved trial design as a result of the group’s dialogue, which included representatives from academia, industry and regulatory authorities from both the United States and Europe [10, 11]. The group’s consensus on the leading cause of failure to convincingly demonstrate safety and efficacy of agents in patients hospitalized with WHF was identified as the lack of in-depth understanding of the molecules before pivotal multi-center international trials were conducted. This is not a problem simply in drug development in patients with WHF, but rather a systematic problem spanning different disease syndromes.
In the current paradigm of drug development, the successful completion of animal model studies is followed by phase I trials, which are focused on the investigation of the safety of novel agents in healthy subjects. This is followed by phase II studies to further understand the mechanistic properties, dose selection, and broad safety assessment in the context of a hypothesis-drive study in the desired target population. In a heterogeneous condition such HF, selection of the right patient population to target in a small phase II trial can be difficult and is critical for both efficacy and safety. This then culminates in a pivotal large-scale phase III trial (Fig. 31.1). Unfortunately, under this schema, every large trial conducted to date in patients hospitalized with WHF has failed to produce positive results in terms of efficacy and/or safety [12–19].
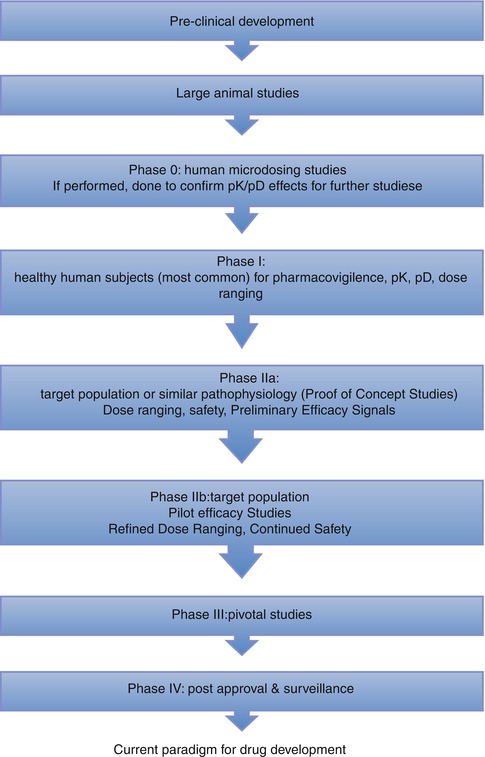
Fig. 31.1
Current Paradigm for Drug Development (Adapted and reproduced from Gheorghiade et al. [21] by permission)
As described in Chap. 1, the Clinical Research Roundtable at the Institute of Medicine outlined 2 main obstacles limiting the transfer of discovery of disease mechanisms at the “bench” from being implemented at the “bedside” as novel methods for diagnosis, therapy, and prevention in the biomedical sciences [20]. These are defined as the T1 Block, translation from basic sciences to human studies, while the T2 block focuses on translation of new knowledge into everyday clinical practice and health care decision-making. Based on the group’s prior experiences in clinical trial design, the main obstacle to overcome in patients hospitalized with WHF was identified as the T1 block. Therefore, we propose a novel paradigm of the T1 model to focus on investigation of the mechanistic effects of experimental agents prior to phase II studies prior to pivotal large-scale testing. We anticipate that this novel paradigm will improve the likelihood of successful translation of the beneficial effects of experimental agents observed in animal models into patients (Fig. 31.2) [21].
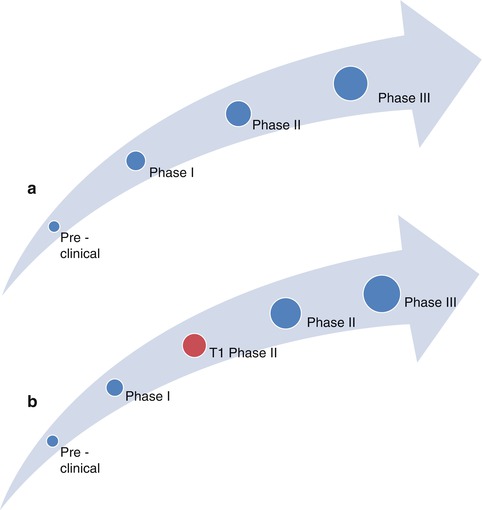
Fig. 31.2
How to adapt the current schema to include a T1 model (Adapted and reproduced from Gheorghiade et al. [21] by permission). The current schema (a) and the proposed paradigm (b) for drug development
31.2.1 T1: Translational Mechanistic Phase of Research
The transition from preclinical studies to clinical trials in the quest for a novel agent developed at the “bench” to be administered at the “bedside” providing tangible human benefit requires the utmost of care. Specifically, the last decade of failed trials in patients with WHF have increased awareness that inadequate attention to detail regarding the mechanism of action of a novel agent and patient selection are recipes for disaster. Examples from recent clinical trials are outlined in Table 31.2. Despite a well-established risk of hypotension, the potential adverse effects of coronary hypoperfusion as a result were not well studied in Levosimendan, especially in patients with concomitant coronary artery disease (CAD) [22]. The negative result in the Value of Endothelin Receptor Inhibition with Tezosentan in Acute Heart Failure Studies [15], while disappointing, was not unexpected as the hemodynamic and mechanistic effects of tezosentan were not previously tested in-depth at the lower dose in phase II trials prior to escalation to a multi-center international phase III trial [15, 23]. Tolvaptan, an oral vasopressin 2 receptor antagonist was developed without complete understanding of the physiologic response, which may be unfavorable, to unopposed vasopressin 1 activity on the heart and vasculature as a result of selective vasopressin 2 inhibition [24]. The promising renal-protective effects of adenosine blocking agents in small early-phase trials prompted the Placebo-Controlled Randomized Study of the Selective A1 Adenosine Receptor Antagonist Rolofylline for Patients Hospitalized with Acute Decompensated Heart Failure and Volume Overload to Assess Treatment Effect on Congestion and Renal Function (PROTECT) without an intermediate study assessing the impact of improved renal function on mortality and morbidity [25]. It was only after a decade of investigation and nearly $1 billion dollars that the Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure (ASCEND-HF) demonstrated lack of clinical efficacy, either in short-term improvement of dyspnea, or 30-days mortality or rehospitalization for heart failure [26].
T1 phase studies will be designed to occur after initial human studies in phase 0 and 1 studies, which will have demonstrated safety to proceed with further drug development and investigation in human subjects. This proposed schema would promote a seamless transition from animal studies to phase II investigations. T1 studies will be designed similar to animal studies with relatively small numbers of homogenous patients to allow for initial hypothesis testing. As we will explore later, the heart failure population is quite heterogeneous and clinical conditions, such as variable background therapy cannot be controlled in a clinical trial, and this may significantly affect results of small early phase studies. Therefore, patients entered into a T1 study will have a common etiology for WHF, a narrow range of age, ejection fraction, background therapy, and kidney function, as these are critical regulators of prognosis and may result in differential responses to novel therapies. This emphasis on homogeneity will be an obstacle in recruitment, however, the relatively small sample size required for the T1 studies will be expected to mitigate the difficulty. Further, investigation of multiple small homogenous groups will allow the ability to best capture which subgroup may benefit the most and should be considered as the initial target population in larger studies.
T1 studies will focus on mechanistic investigation of the novel therapeutic agent in the pre-specified patient population. These studies will focus on clearly defined end-points (e.g. changes in pulmonary capillary wedge pressure, renal function), but not on outcomes, as the small sample size would be prohibitive. This will allow for comprehensive initial investigation targeting the new drug’s effect specifically on the heart. The advances in the past decade in biomarkers and cardiac imaging, such as 3-D echocardiography and cardiac magnetic resonance imaging (CMRI) make this phase possible today by allowing us the ability to assess, not only the left ventricular gross function, but also the myocyte, the interstitium, and the metabolism of the heart. This sets the stage for a novel characterization of HF patients to assist in enrollment of a more homogenous patient population. Experts in these imaging areas as well as electrophysiology, angiography, nephrology, and clinical trialists, will need to be identified up-front for successful trial design and execution. A small consortium of experienced centers is currently being created to conduct these trials with experience and expertise in these fields, especially in novel echocardiography techniques (tissue Doppler imaging, strain rate analysis, three-dimensional image acquisition), novel imaging modalities with cardiac magnetic resonance imaging and spectroscopy to evaluate myocardial metabolism, and invasive hemodynamics with right heart catheterization. In addition to standardization and optimization of the quality of hemodynamic assessment and image acquisition and interpretation, the use of concomitant “standard” background therapy has significant variability in large clinical trials as it is often left up to the discretion of the individual investigator or treating physician. The use of select T1 centers with a pre-specified protocol for initiation and up-titration of “standard” evidence-based background therapy will be critical to minimize variability in this regard. Furthermore, this may help attenuate the continental differences that were observed in the EVEREST Tolvaptan trial [27].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


