Fig. 24.1
The embryonic heart just after cardiac looping. The common atrium (CA) communicates with the left ventricle (LV) only, via the atrioventricular canal (AVC). The primary ring (PR) marks the boundary between the left ventricle, derived from the first heart field (straight heart tube), and the developing right ventricle (RV), derived from the anterior part of the second heart field. The outflow tract (OT) is entirely above the developing right ventricle
Muscular defects
Defects opening into the outlet of the ventricles (outlet defects)
Defects opening into the inlet of the ventricles (inlet defects)
Central membranous defects
The anatomic characteristics of these defects and their assumed embryologic origin are depicted in Table 24.1.
Table 24.1
The four main anatomic types of ventricular septal defect with their anatomic characteristics and their presumably embryologic origin
Type of defect | Fibrous continuity | Embryologic origin | Time in cardiac development | ||
|---|---|---|---|---|---|
Muscular | Midmuscular | NO | Compaction of myocardium, trabeculations | T | |
Apical | |||||
Inlet muscular | |||||
Central membranous without aortic overriding | YES (septal tricuspid leaflet/aortic leaflets) | Formation of membranous septum (AV cushions) | T-1 | ||
Outflow tract (outlet) defects | Malalignment defects (aortic overriding) | With aortic-tricuspid fibrous continuity (perimembranous extension) | YES (anterior tricuspid leaflet/aortic leaflets) | Cardiac neural crest and anterior second heart field. Outflow tract cushions | T-2 |
Muscular borders | NO (muscular borders) | ||||
Juxta-arterial defects (fibrous continuity Ao/pulm valves) | With aortic-tricuspid fibrous continuity (perimembranous extension) | YES (anterior tricuspid leaflet/aortic leaflets) | |||
Muscular borders | NO (muscular borders) | ||||
Inflow tract (inlet) defects | Common atrioventricular junction (isolated ventricular component) | YES (tricuspid/mitral) | Posterior second heart field. AV cushions | T-3 | |
Straddling tricuspid valve (malalignment atrial/ventricular septum) | Convergence. Formation of AV junction | T-4 | |||
Ventricular septation, as a crucial step in heart development, was studied first in human embryos from a purely morphologic point of view, then on chick and mouse. Recent techniques such as high-resolution episcopic microscopy and high-resolution magnetic resonance imaging (MRI) have been used in mouse and human embryos to better analyze the development of the atrial and ventricular septa [4, 5]. Despite these recent advances, the precise mechanisms leading to normal ventricular septation are not fully understood and controversies still exist. Some of them are due to some developmental differences in the chick and in the mouse. On the other hand, despite some minor differences, the steps of cardiac development are very similar in mouse and human [6].
24.2 Normal Ventricular Septation
Ventricular septation is a complex process starting from the onset of cardiac looping to its final steps which are the formation of the membranous septum and later on, during fetal life, compaction of the ventricular myocardium.
Immediately after cardiac looping (early looping stage), the inlet segment (atria and atrioventricular canal) is located entirely above the future left ventricle, and the outlet segment (the conotruncus or outflow tract) is located entirely above the future right ventricle, leading to both double-inlet left ventricle and double-outlet right ventricle types of atrioventricular (AV) and ventriculo-arterial connection (Fig. 24.2). From this stage on, the heart continues to grow by addition of myocardial cells from two cellular populations: the first heart field or primitive linear heart tube will form the left ventricle and a small part of the atria above the AV valves; the second heart field (SHF) is essential for the development of the right ventricle and the outflow tract (OFT) (anterior SHF) and also the venous pole, including the major part of the atria (posterior SHF or dorsal mesocardium).
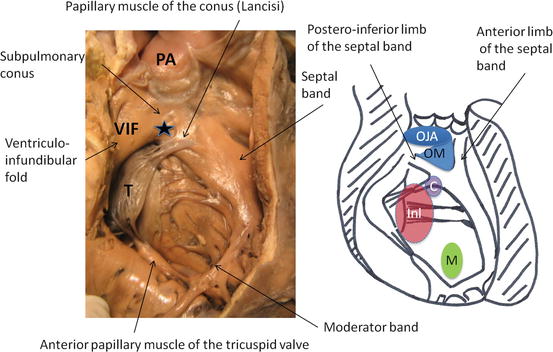
Fig. 24.2
The right ventricle in the normal heart, and the four main anatomic types of ventricular septal defect, viewed from the right ventricle. Asterisk outlet septum, C central membranous, Inl inlet, OJA outlet, juxta-arterial, OM outlet, malalignment, PA pulmonary artery, M muscular, T tricuspid valve, VIF ventriculo-infundibular fold
Ventricular septation involves not only myocardial cells of the two heart fields, but also mesenchymal cells of the endocardial cushions; most of those will be secondarily muscularized. Proper ventricular septation can be achieved normally only if all elements of the definitive four-chambered heart are correctly aligned.
The interventricular septum first appears immediately after the cardiac loop, as a myocardial ridge developed at the posterior part and at the apex of the primitive heart, the primary ring (Fig. 24.2). Cardiac looping creates an outer curvature, formed by the ventral surface of the straight heart tube, and an inner curvature, between the outflow and the inflow parts of the heart. The outer curvature will take part to the growth of the ventricles, by ballooning of the cavities with formation of multiple trabeculations [7], while the remodeling of the inner curvature controls the alignment between the inlet and outlet segments [8]. The right ventricle develops later than the left ventricle [9], by addition of the cardiomyocytes of the anterior SHF [10]. This differential growth contributes to the alignment of the developing atrial and ventricular septa. The formation of the interventricular septum would thus be both passive (ballooning) and active [11–13].
The ventricular septum can be divided in several components, each one having a different embryologic origin. The muscular trabeculated septum is composed of the myocytes of both left and right ventricles [11] and thus originates from both first and second heart fields.
The origin of the inlet septum is still controversial. What is commonly called the inlet septum is the posteroinferior, or caudal, part of the ventricular septum [12, 14], but for some authors it does not exist in the normal heart [4, 15]. Experimental studies in the mouse embryo demonstrate that the larger part of the central mesenchymal mass created by the fusion of the mesenchymal tissues of the vestibular spine, the mesenchymal cap of the primitive atrial septum, and the AV cushions, becomes muscularized to form the inferior margin of the oval fossa [16]. Whether muscularization occurs or not in the posteroinferior part of the mass (the part which is lacking in hearts with a common AV junction, forming the inlet VSD) has been demonstrated in the chick embryo [17] but not in mammals, in which the posterior-inferior part of the ventricular septum would be part of the developing trabeculated muscular septum [18].
The outlet septum develops by fusion and muscularization of the proximal part of the endocardial cushions of the OFT [19] and is taken along leftward by the rotation of the developing aortic valve, to fuse with the upper primitive ventricular septum between the two limbs of the septal band or septomarginal trabeculation (Fig. 24.3), itself derived from the primary fold [20]. This rotation depends on the proper growth of the OFT by addition of myocardial cells from the anterior heart field, in response to the migration of cardiac neural crest cells toward the OFT [21]. The rotation of the aortic valve permits its transfer toward the left ventricle and its wedging between the tricuspid and mitral valves, establishing fibrous mitral-aortic continuity [22–24].
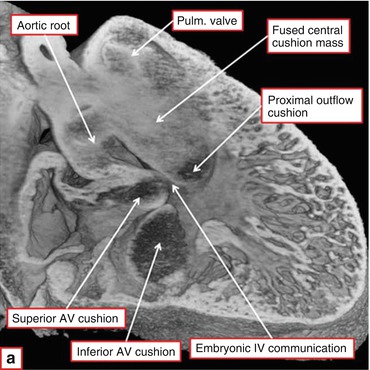
Fig. 24.3
Episcopic section from a developing mouse heart at E13.5. The aortic root is still above the right ventricle. The proximal outflow cushions have fused but have not yet muscularized. The embryonic interventricular communication lies at the confluence of the outflow tract cushions and the superior and inferior atrioventricular cushions (Courtesy of R.H. Anderson)
The membranous septum (Fig. 24.4) completes cardiac septation by closing the residual central defect, by fusion of the AV canal cushions with the left outflow tract cushion [2]. Any failure of development of these different parts (the limits of which are barely recognizable in the normal heart) leads to a corresponding phenotype of VSD.
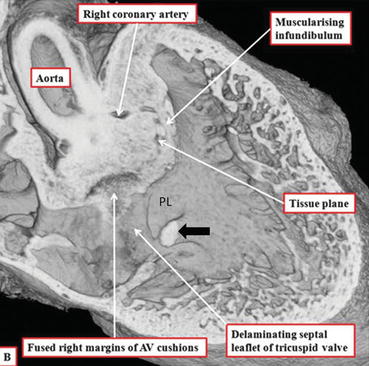
Fig. 24.4
Episcopic section from a developing mouse heart at E14.5. The fused central cushion mass is inserted between the two limbs of the septomarginal trabeculation (septal band) and has begun to muscularize. It will be transformed into the outlet septum and into the free-standing muscular subpulmonary infundibulum. The aorta has achieved its transfer toward the left ventricle. The rightward margins of the atrioventricular endocardial cushions fuse to close the right ventricular entrance to the subaortic vestibule, completing the process of ventricular septation. The membranous septum which results from this fusion is visible under the posterior limb (PL) of the septal band (black arrow) (Courtesy of R.H. Anderson)
24.3 Pathways to the Different Anatomic Types of Ventricular Septal Defects
The molecular determinants of the complex events leading to ventricular septation are not fully understood yet [25]. This is due in part to the uncertainties relative to the normal development of the ventricular septum and also to the difficulties in assessing the precise anatomic phenotype in animal models. In addition, few animal models exhibit isolated VSDs.
A lot of genes coding for transcription factors have been recognized as playing a role in ventricular septation [26, 27]. However, the major ones, T-box 5 (Tbx5), T-box 20 (Tbx20), GATA binding protein 4 (Gata4), GATA binding protein 6 (Gata6), and NK2 homeobox 5 (Nkx2-5), when deleted in mouse or mutated in human, produce VSDs but also atrial septal defects (ASDs) and atrioventricular septal defects (AVSD) (see Chaps. 20 and 26). An explanation for this could be that several genes act on cells from both parts of SHF, anterior and posterior. For example, sonic hedgehog (Shh) signaling has been found essential for the contribution of the anterior heart field to the developing outflow tract in mice [28] and regulates the expression of Tbx1 and the semaphorin receptor neuropilin 2 in the SHF [29]. Shh signaling has also been demonstrated to play a crucial role in the formation of the vestibular spine, which originates from the posterior SHF, loss of Shh from the pharyngeal endoderm resulting in AVSD in mice [30].
24.3.1 Pathways to the Outlet VSDs
All outflow tract defects share the same anatomic type of VSD, opening to the outlet of the right ventricle and located between the two limbs of the septal band (except transposition of the great arteries, in which this type of VSD is not constant, and double-outlet right ventricle (DORV) with noncommitted VSD) [31]. The phenotypic differences observed, especially regarding the posteroinferior rim of the VSD (fibrous, with fibrous continuity between the anterior leaflet of the tricuspid valve and the aortic valve, or muscular, due to the fusion of the posterior limb of the septal band with the ventriculo-infundibular fold), are related to the degree of rotation of the aortic valve during heart development [31]. Separation of the posterior limb of the septal band from the ventriculo-infundibular fold (so-called outlet VSD with perimembranous extension) would indicate a higher degree of rotation of the aortic valve, by analogy with the normal heart [31]. Therefore, the molecular pathways that lead to an outlet VSD have to be the same than those leading to so-called conotruncal, or cardiac neural crest, defects, which include tetralogy of Fallot and its anatomic variants, common arterial trunk, interrupted aortic arch type B, isolated outlet VSDs, and DORV with outlet VSD. The genetic background of these anomalies is well known, because of their frequent association with 22q11 microdeletion or DiGeorge syndrome (see Chaps. 23 and 32). The major candidate gene for DiGeorge syndrome is Tbx1, which is expressed in the anterior part of the SHF [29] and is a key regulator of SHF development. Tbx1+/− mice have defects of the aortic arches (especially the 4th aortic arch) and great arteries, while Tbx1−/− mice have outflow tract defects, with variable severity according to the dosage of the gene [32]. This dosage effect, interacting with the complex molecular pathways affecting the anterior SHF, could explain the phenotypic differences observed between the various outflow tract defects and within the same malformation. Other animal experiments have shown that Tbx1 is expressed in a particular SHF subdomain, which contributes to the subpulmonary myocardium [29]. In Tbx1 mutant mice, the outflow tract is shorter and narrower, due to reduction in size of the future subpulmonary myocardial domain [33].
24.3.1.1 Malalignment-Type Outlet VSDs
These defects are due to a malalignment between the developing outflow tract and the ventricles, due to lack of rotation of the aortic valve, and result in a failure of fusion of the outlet septum with the primitive ventricular septum between the two limbs of the septal band. This is due to a lack of addition of myocardial cells by the anterior SHF, in interaction with neural crest cells, to the developing outflow tract [34], resulting in an underdevelopment of the subpulmonary myocardium [29]. All the genes involved in regulation of anterior SHF, when mutated in animals and in humans, can determine, in addition to so-called conotruncal defects, isolated malalignment outlet VSDs.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


