Class
Genes
Transcription factor
Cited2, Foxp1, Gata3, Gata4; Gat6, Gata6, Jun, Msx1; Msx2, Pax3, Pbx1, Prdm1, Prdm3, Sox4, Tbx1, Tcfap2a
Signalling molecule
Rara; Rarb, Rara; Rarc, Rxra, Sema3c, Shh
Growth factor
Fgf8, Vegfa
Protein binding
Bmp4, Chrd, Edn1, Ednra, Flna, Ltbp1L, Pinch1
Receptor
Acvr1, Bmpr2, Pdgfra, Plxnd1, Smo, Tgfbr1, Tgfbr2
Enzyme
Aldh1a2, Ece1
48.2 T-Box 1
Malformations of the great arteries are a major feature in 22q11 deletion syndrome (22q11DS, also known as DiGeorge, velocardiofacial or Shprintzen syndrome). The transcription factor T-Box 1 (TBX1) is thought to be the main causative gene for the 22q11DS phenotype [3–5] and several animal models, including mouse and zebrafish, with mutant Tbx1 alleles have been created, allowing this gene to be studied in great depth to understand its role in the formation and patterning of the cardiovascular system, including the pharyngeal arch arteries (see Chap. 42).
Transgenic mice null for Tbx1 have a severe phenotype, and they die in the perinatal period with cardiovascular defects including CAT (Fig. 48.1). Although this phenotype is assumed to be caused by a failure of neural crest cells to properly septate the outflow tract, Tbx1 protein is not expressed within the neural crest itself. It is therefore believed that loss of Tbx1 affects neural crest cell migration [7–9].
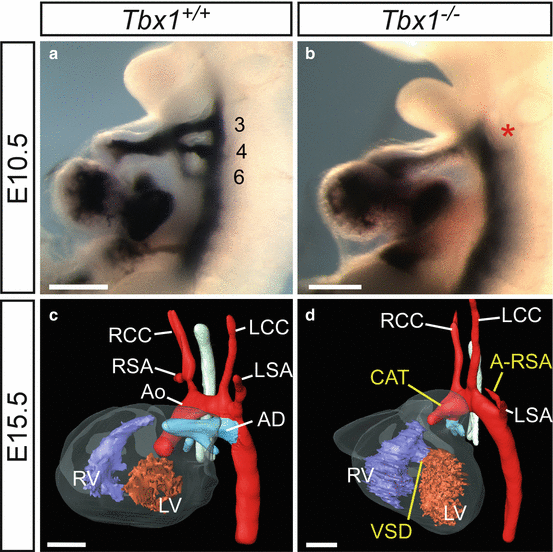
Fig. 48.1
Cardiovascular defects in Tbx1-null (Tbx1 −/−) mouse embryos and foetuses. (a, b) Injection of India ink into the heart of E10.5 embryos. (a) In the wild-type embryo (Tbx1 +/+) three bilaterally symmetrical PAA are present (numbered 3, 4 and 6) which are fully patent to ink. (b) In the Tbx1 −/− embryo a single outflow vessel is present (asterisk) revealing that the PAA have failed to form properly. (c, d) 3-D reconstructions from MRI datasets [6] of E15.5 foetuses. (c) By E15.5 the heart and its associated great vessels have developed into the mature configuration. (d) The heart of a Tbx1 −/− foetus presents with great artery defects related to the failure of the PAA to form correctly: CAT and A-RSA as well a VSD. The heart in this foetus is abnormally rotated to the left. Abbreviations: Ao aorta, AD arterial duct, LCC left common carotid, LSA left subclavian artery, LV left ventricle, RCC right common carotid, RSA right subclavian artery, RV right ventricle. Scale, 500 μm
Genetic modification of mice can be used to conditionally inactivate a gene in a temporal and spatial manner. When this is performed with Tbx1, the timing of expression within the developing embryo, and the consequences this has on cardiovascular development, can be ascertained. For example, when Tbx1 is inactivated between E7.5 and E8.5 of embryonic development, PAA defects occur such as interruption of the aortic arch (IAA) and aberrant right subclavian artery (A-RSA), but later deletion between E8.5 and E9.5 affects outflow tract septation, resulting in CAT [10]. Regulation of Tbx1 gene dosage is also critical. Tbx1 mRNA dosage levels can be controlled through a series of mutant Tbx1 alleles in transgenic mice [11]. These hypomorphic alleles reveal that a critical threshold of Tbx1 mRNA is required for correct formation of the PAA. A reduction in Tbx1 mRNA levels to ~20 % of wild type was sufficient to introduce the CAT phenotype. The overexpression of Tbx1 in transgenic mice also results in cardiovascular abnormalities, including CAT and IAA [12].
As the Tbx1-null phenotype affects a wide range of tissues, including many of the embryonic structures derived from the pharyngeal arches and second heart field, including the outflow tract, interventricular septum, PAA, thymus, parathyroids, ears and craniofacial development (reviewed in [13, 14]), it is possible that each expression domain may contribute to different aspects of the phenotype. Therefore, conditional deletions of Tbx1 have been used to investigate its tissue-specific requirements in the context of Tbx1-null embryos. Conditional deletion of Tbx1 within the pharyngeal endoderm, ectoderm and mesoderm in addition to the SHF through the use of Nkx2–5Cre resulted in full recapitulation of the CAT and ventricular septal defect (VSD) phenotypes observed in Tbx1 −/− mutants [15]. A reduced and disorganised smooth muscle cell layer surrounding the outflow tract was also observed, possibly due to the reduced proliferative activity in the second heart field [15]. Mesoderm posterior 1 homolog/Cre-recombinase (Mesp1Cre)-mediated deletion of Tbx1, which removed the gene from all mesodermal derivatives, including the endothelium, also resulted in 100 % penetrance of CAT, VSD and severe PAA malformations [9]. In addition, the segmentation of the pharynx is disrupted, resulting in the loss of the caudal pharyngeal arches, as is observed in Tbx1 −/− mutant mice. Further defects affecting the migration of the NCCs into the pharyngeal arches, disruptions of the neural crest-derived cranial ganglia and reduced proliferation of the neural crest-derived pharyngeal arch mesenchyme were also observed. Reactivation of Tbx1 within the mesoderm of mutant embryos rescued the outflow tract and VSD phenotypes, but not the 4th PAA defects, suggesting other tissues are required in this aspect of the phenotype [9], most likely the epithelium.
Other animal models have been employed to study the role of Tbx1 in cardiovascular development, and Tbx1 is expressed throughout the pharyngeal regions of the frog [16], chicken [17] and zebrafish [18]. In the chicken, blocking cytochrome P450, family 26, subfamily A, polypeptide 1 (Cyp26a1), a gene required for retinoic acid inactivation during embryogenesis, results in outflow tract defects, i.e. CAT and double-outlet right ventricle (DORV), and PAA defects similar to that seen in the mouse model of Tbx1 loss [17]. Tbx1 levels were downregulated, and the retinoic acid-synthesising enzyme retinaldehyde dehydrogenase 2 (Raldh2) was upregulated. Consistent with a role for sonic hedgehog (Shh) in regulation of Tbx1, chick embryos exposed to Shh-soaked beads responded by upregulating Tbx1 expression in the pharyngeal arches [19].
48.2.1 Fibroblast Growth Factors
Mice expressing a hypomorphic allele of fibroblast growth factor 8 (Fgf8) present with outflow tract defects such as CAT and DORV [20, 21]. The Fgf8 receptors, FGFR1 and FGFR3, are expressed within neural crest cells and are thought to be required in neural crest cell migration. Plating neural crest cells in the presence of Fgf8 in vitro increased their migratory capacity, while inhibition of FGFR1 both in vitro and in vivo reduced migration [22], indicating that this pathway may be required in neural crest cell migration and survival within the pharyngeal arches. A reduction in Fgf8 signalling may therefore result in a decreased number of neural crest cells entering the outflow tract, thereby causing DORV, while a more severe reduction in the number of cells of this type would result in CAT [22].
48.2.2 Transforming Growth Factors
The signalling molecule transforming growth factor (TGF)-β is vital for regulating many basic cell responses and has been shown to be important for the development of the great arteries in mouse models (see Chap. 36). The TGFβ signalling pathway involves the binding of the TGFβ ligand to the cell surface TGFβ type II receptor (TGFBR2), which subsequently activates the TGFβ type I receptor, TGFBR1 (also known as ALK5). This results in the phosphorylation of SMAD family member protein (SMAD) 2 and SMAD3 proteins, binding to SMAD4 and translocation to the nucleus to regulate downstream gene expression. Transgenic mice null for the TGFβ2 ligand (Tgfb2) die perinatally with cardiovascular defects, which includes CAT [23, 24]. The TGFβ receptors Tgfbr1 and Tgfbr2 are essential for embryogenesis as mice lacking these genes die around mid-gestation [25, 26]. Conditional alleles, however, allow for this early embryonic lethality to be avoided and the role of these genes in cardiovascular development dissected. When the Tgfbr1 and Tgfbr2 receptors are deleted from the neural crest using wingless-type MMTV integration site family, member 1 (Wnt1)-Cre transgenic mice, the embryos present with CAT [27–29]. This suggests that TGFβ signalling is required for neural crest cells to control septation of the aorta and pulmonary trunk. CAT is also seen in embryos when the long form of latent TGFβ binding protein 1 gene (Ltbp1) is disrupted [30].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


