Fig. 33.1
Schema illustrating the progressive developmental steps during formation and remodeling of the outflow tract (OFT), together with the major cell types and developmental processes involved. HT heart, AP arterial pole, VP venous pole, SHF second heart field, PAA pharyngeal arch artery, RA right atrium, LA left atrium, RV right ventricle, LV left ventricle, Ao aorta, AoV aortic valve, DA ductus arteriosus, PT pulmonary trunk, PV pulmonary valve, SPMc subpulmonary myocardium, IVS interventricular septum
33.2 TOF and DORV are Part of a Spectrum of Outflow Tract Alignment Defects
Addition of cardiac progenitor cells from the SHF in pharyngeal mesoderm to the elongating poles of the heart drives heart tube elongation during looping morphogenesis [1]. The entire OFT is derived from the SHF, and direct or indirect impairment of SHF deployment results in OFT shortening [2]. Maximal OFT extension creates the template for subsequent septation and is critical for correct ventriculoarterial alignment. Septation is driven by coincident cushion morphogenesis and influx of neural crest-derived mesenchyme that together divide the OFT into the ascending aorta and pulmonary trunk [3]. During this process, the OFT rotates in a counterclockwise direction (with respect to the direction of blood flow), and the future pulmonary trunk is positioned in a ventral position above the right ventricle, the ascending aorta becoming wedged between the atrioventricular valves and aligned with the left ventricle (Fig. 33.2). Extensive evidence suggests that septation and alignment are distinct, though coincident, processes: failure of OFT septation will result in common arterial trunk (or persistent truncus arteriosus), while failure of alignment will result in a spectrum of alignment defects, ranging from mild to severe [4, 5]. At the mild end of the spectrum, failure of the aorta to make an exclusive connection with the left ventricle results in overriding aorta (OA), where the aorta connects with both ventricles above a membranous VSD. The VSD arises due to malalignment of the ventricular and outflow tract septa. When associated with pulmonary stenosis or atresia, overriding aorta results in a TOF phenotype. If >50 % of the aorta lies over the right ventricle, overriding aorta becomes DORV, with a subaortic VSD. Total failure of OFT rotation, in the absence of a leftward shift of the OFT, results in a dextroposed aorta that connects with the right ventricle in a side-by-side configuration with the pulmonary trunk (Fig. 33.2). This defect, DORV with a subpulmonary VSD, is known as Taussig-Bing syndrome and is a configuration related hemodynamically to TGA. TOF and DORV thus lie within a disease spectrum of alignment defects and are distinguished by the degree of aortic dextroposition.
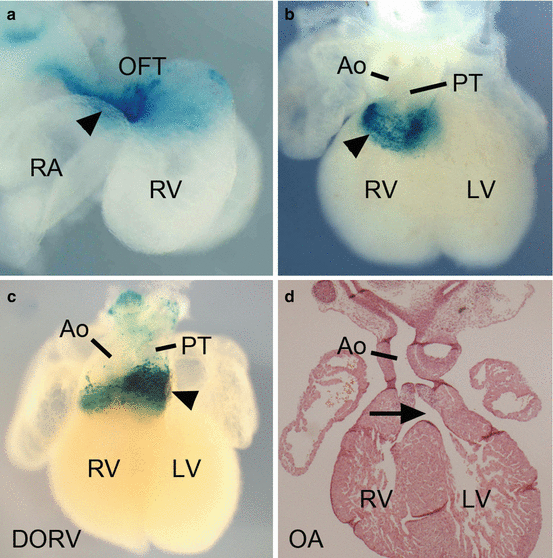
Fig. 33.2
Mouse hearts illustrating rotation of the myocardial OFT wall using a transgene enhancer trap (line y96-Myf5-nlacZ-16) expressed in the inferior wall of the midgestation OFT (a) and in subpulmonary myocardium after septation (b). Transgene-driven β-galactosidase activity is visible (arrowheads) in blue after incubation in X-gal solution. Expression of the y96-Myf5-nlacZ-16 transgene in a fetal Tbx3 −/− heart (c) with double outlet right ventricle (DORV) and a leftward positioned pulmonary trunk (arrowhead). Histological section through an Fgfr2IIIb −/− heart (d) with overriding aorta (OA), showing the aorta aligned with a ventricular septal defect (arrow). RA right atrium, RV right ventricle, OFT outflow tract, Ao aorta, PT pulmonary trunk, LV left ventricle
33.3 The Second Heart Field and Outflow Tract Elongation
Failure to maximally elongate the embryonic OFT results in an OFT that is too short to make an exclusive connection between the ascending aorta and left ventricle. Defects in SHF progenitor cell deployment range from defects in addition of cells to the distal OFT to total failure to extend the linear heart tube, producing a consequent spectrum of mild to severe OFT defects, including ventriculoarterial alignment defects [2, 6]. Addition of cells giving rise to myocardium at the base of the pulmonary trunk, known as subpulmonary (or infundibular) myocardium, is of particular importance for alignment defects. Indeed, underdevelopment of subpulmonary myocardium is thought to be a primary cause of TOF [7]. Ablation of the SHF in chick embryos results in a reduction of subpulmonary myocardium and OA with pulmonary stenosis (TOF) [8]. Future subpulmonary myocardium is the last myocardial derivative of the SHF to be added to the OFT, giving rise to the inferior OFT wall [9]. Subpulmonary myocardium originates in the posterior SHF, adjacent to progenitor cells that contribute to the venous pole of the heart and are critical for atrioventricular septation [10]. Clonal analysis and lineage experiments have shown that subpulmonary and venous pole myocardium are clonally related, identifying a common progenitor cell population in the SHF that segregates to the cardiac poles to give rise to parts of the heart affected in common forms of CHD [11–13].
Many transcriptional regulators and signaling pathways have been implicated in the control of proliferation and progressive differentiation of SHF progenitor cells [2, 14]. In mouse embryos, addition of subpulmonary myocardium is dependent on the T-box containing transcription factor Tbx1, encoded by the major gene implicated in del22q11.2 (or DiGeorge) syndrome [15]. Haploinsufficiency for TBX1 in human patients is the most common known genetic cause of TOF, accounting for 15 % of TOF [6, 16]. Tbx1 is required for proliferation and delayed differentiation in the SHF [17, 18]. Whereas homozygous Tbx1 null mouse embryos have common arterial trunk with failure of subpulmonary myocardial addition, mice carrying hypomorphic Tbx1 alleles with <25 % of normal Tbx1 levels have a range of alignment defects including DORV and OA [19]. Among other transcriptional regulators of SHF development, NK2 homeobox 5 (Nkx2-5) is essential for elongation of the early heart tube. As in the case of Tbx1, hypomorphic embryos with <25 % of normal Nkx2-5 levels display alignment defects including DORV and OA, preceded by a shortened OFT and decreased proliferation in the SHF [20]. Embryos doubly heterozygous for GATA binding protein (Gata) 4 or Gata6 plus Gata5 also display DORV and overriding aorta [21]. Gata3 mutant embryos also have a shortened OFT and DORV and common arterial trunk phenotypes [22]. Loss of function of the Gata co-factor friend of GATA 2 (Fog2; or zinc finger protein, FOG family member 2; ZFPM2), or the domain of Gata4 that interacts with Fog2, also results in CHD, including OA and DORV phenotypes [23, 24]. Mutation of homeobox A1 (Hoxa1), expressed in subpulmonary myocardial progenitors in the SHF, has recently been shown to result in an OA phenotype [11, 25].
Intercellular signaling pathways play an important role in SHF development. Fibroblast growth factor (FGF) signaling controls SHF proliferation downstream of Tbx1 [16]. Mouse embryos lacking components of the FGF signaling pathway display OFT shortening and alignment defects, including Fgf receptor Fgfr2IIIb null embryos, mesodermal conditional Fgfr1 and Fgfr2 null embryos, and Fgf8 hypomorphic and mesodermal conditional mutant embryos [26–29]. The incidence of alignment defects in mesodermal Fgf8 mutant embryos is increased when alleles of Fgf10 are additionally removed [30]. Fgf15 null embryos also display DORV and OA [31]. In chick embryos, treatment with Fgf8-blocking antibodies or an FGF receptor antagonist results in DORV and OA phenotypes with pulmonary stenosis or atresia [32]. Additional intercellular signaling pathways regulating SHF development include Notch signaling: conditional Notch mutant embryos and embryos lacking the Notch target genes hes family bHLH transcription factor 1 (Hes1) and hes-related family bHLH transcription factor with YRPW motif 1 (Hey1) display OFT shortening and DORV and OA with pulmonary stenosis and are associated with impaired FGF signaling and proliferation in the SHF [33–35]. Deletion of Jag1 in the SHF results in DORV (in addition to common arterial trunk and atrial septal defects) and jagged 1 (Jag1) Notch2 compound heterozygous embryos display OA with pulmonary stenosis [35, 36]. Endothelial specific Jag1 deletion also results in OA, highlighting the multiplicity of roles different intercellular signaling pathways play during OFT development [37]. DORV is also observed in embryos lacking an enzyme encoded by the gene presenilin 1 (Psen1), required to activate the Notch receptor [38].
Proliferation in the SHF is also regulated by canonical Wnt and hedgehog signaling pathways [2, 14]. Indeed, mouse embryos cultured in the presence of an activator of the canonical Wnt signaling pathway have elongated OFTs and expanded numbers of SHF cells; conditional loss of function of canonical Wnt signaling leads to a reduction in progenitor cell numbers [39]. Chick embryos cultured in the presence of the Shh antagonist cyclopamine have decreased proliferation in the SHF resulting in OA [40]. Genetic tracing in the mouse has shown that SHF cells exposed to Shh signaling contribute to subpulmonary myocardium [41]. Mouse embryos lacking Shh display a single ventricular outlet, corresponding to total pulmonary atresia [42]. Retinoic acid signaling is also active in future subpulmonary myocardial progenitor cells. Analysis of retinoic acid receptor mutant embryos have revealed a role in renewal of SHF cells during OFT elongation leading to DORV and OA phenotypes [43]. Exposure of mouse embryos to retinoic acid also results in DORV (together with common trunk and TGA) phenotypes, revealing critical RA dosage requirements for SHF deployment [44, 45]. The planar cell polarity pathway regulates polarized cell movements and has also been implicated in the regulation of SHF deployment and OFT alignment [46]. Mouse embryos lacking components of this pathway encoded by genes such as dishevelled segment polarity protein 2 (Dvl2), VANGL planar cell polarity protein 2 (Vangl2), or Scribble (Scrb) have shorter OFTs and consequent alignment defects [46–48]. Double heterozygous Vangl2 +/− ;Scrb +/− mutant mice also have DORV [49]. Planar cell polarity plays an important role in epithelial SHF cells during their deployment to the elongating OFT [47]. Vangl2 mutant mice display altered epithelial properties in the distal OFT during myocardial differentiation that may be essential for maximal OFT elongation [48]. The noncanonical wingless-type MMTV integration site family (Wnt) ligand encoding genes Wnt5a and Wnt11 have been implicated upstream of PCP during SHF deployment and OFT morphogenesis [47, 50, 51]. Embryos lacking either of these genes display DORV, as well as common arterial trunk and TGA phenotypes; double mutant mice have a severe reduction of SHF progenitors [52]. Wnt5a is a direct target of Tbx1 in the SHF and has been shown to phenocopy the subpulmonary myocardial deficit of Tbx1 null embryos [53, 54].
Intercellular signals controlling SHF development are both autocrine in origin, as in the case of Notch and FGF signaling, and from surrounding cell types in the pharyngeal region, including pharyngeal endoderm and ectoderm and neural crest-derived mesenchyme [2, 14]. Together these signals define and regulate the niche of SHF progenitor cells during heart tube extension. Experiments in chick embryos have revealed the critical requirement for neural crest-derived cells in SHF deployment. Ablation of the cardiac neural crest results not only in common arterial trunk due to failure of formation of the outflow tract septum but also to alignment defects such as TOF and DORV. These arise due to perturbation of critical signal exchange between crest and SHF cells in the pharyngeal region prior to the addition of these cell populations to the heart [4]. Neural crest cells have been shown to modulate FGF signaling in the pharyngeal region, thus decreasing SHF proliferation during the terminal stages of heart tube elongation [32]. Neural crest ablation rescues the alignment defects observed in embryos cultured in the presence of Fgf8-blocking antibodies [32]. The precise regulation of proliferation in the SHF is a critical step in progenitor cell deployment, and loss of cardiac neural crest cells perturbs the balance between proliferation and differentiation leading to decreased SHF incorporation and a shorter OFT that fails to wedge and align correctly [5]. Consistent with these findings, mutations in genes affecting cardiac neural crest development in the mouse result in a spectrum of OFT defects that include both septation (direct) and alignment (indirect) defects. Mouse embryos lacking paired box 3 (Pax3), expressed early in the neural crest lineage, for example, display a range of OFT defects including septation defects (common arterial trunk) and alignment defects (DORV) [55]. Mice lacking Tbx3, expressed in different cell types in the pharyngeal region including neural crest-derived mesenchyme, also have a high incidence of DORV [56, 57]. Mutation of pre-B-cell leukemia homeobox (Pbx) 1–3 result in OA, possibly through Pax3 regulation [58, 59]. Together these experiments reveal how perturbation of any of a large number of convergent pathways leads to OFT alignment defects through impaired SHF deployment.
33.4 Outflow Tract Remodeling and Alignment
OFT elongation is clearly not the only prerequisite for correct ventriculoarterial alignment. Maximal elongation of the OFT by addition of SHF progenitor cells creates the template for a series of coincident processes that remodel the OFT into separate right and left ventricular outlets. These processes include OFT rotation, cushion morphogenesis, and differential growth and cell death. Evidence from analysis of developing mouse, chick, and human hearts has shown that the OFT wall rotates in a counterclockwise direction during the septation process and that this is essential for the correct positioning of the great arteries [60–63]. Maximal OFT elongation appears to be required for normal OFT rotation to occur. During the rotation process, the myocardial OFT gives rise to subaortic and subpulmonary myocardium, derived from the superior and inferior OFT walls, respectively [64]. Subpulmonary myocardium assumes a ventral position at the base of the pulmonary trunk (Fig. 33.2). Different degrees of perturbation of OFT rotation result in arrest of this process and lead to positioning of the aortic valve over the right ventricle or to the right of the pulmonary valve, generating alignment defects including OA, DORV, and TGA.
Counterclockwise OFT rotation is driven by the embryonic left-right laterality pathway. Left-right laterality is initiated in the early embryo by cilia-driven flow of signaling molecules to the left side of the node at the anterior end of the primitive streak [65]. Mutation of genes involved in cilia morphogenesis and function result in a range of CHD including alignment defects. These include dynein, axonemal, heavy chain 11 (Dnah11), encoding a dynein molecule mutant in iv/iv mice, together with inv mutant mice, which display DORV, OA, and TGA [66, 67]. Cbp/p300-interacting transactivator, with Glu/Asp-rich carboxy-terminal domain, 2 (Cited2) also regulates early establishment of laterality, and loss of function leads to a range of laterality and alignment defects including DORV and TGA, together with common arterial trunk [68, 69]. Genes involved in ciliogenesis have been identified from mutation screens in mice with alignment defects and TOF patients [70, 71]. Conditional mutagenesis will reveal the extent to which cilia also contribute to alignment defects through later roles in SHF deployment or in flow detection in the developing heart. The failure to establish embryonic laterality results in heterotaxy phenotypes by which independent laterality decisions are made by different components of the heart, leading to a large spectrum of CHD including alignment defects. Asymmetric growth factor signaling at the node establishes distinct left and right domains of gene expression in lateral mesoderm, including left-sided expression of genes encoding the Nodal ligand and the transcription factor paired-like homeodomain 2 (Pitx2) [65]. Pitx2 confers laterality to organs that develop asymmetrically, including the heart. Although not the driver of rightward looping, Pitx2 is expressed in the left side of the SHF and controls OFT rotation [72]. Indeed, Pitx2 mutant embryos are characterized by DORV and TGA, together with common trunk, and subpulmonary myocardium is malpositioned in the absence of Pitx2 [62]. A recent study suggests that the terminal stages of SHF addition are asymmetric and that left-sided addition of subpulmonary myocardial progenitors expressing Nkx2-5 may be a driving force in OFT rotation, a process termed the pulmonary-push model [73]. This would promote counterclockwise rotation of the pulmonary trunk around the aorta. Interestingly, this process is impaired in embryos lacking the Vegf164 isoform: insufficient addition of subpulmonary myocardium in these embryos is likely to account for subsequent DORV and pulmonary hypoplasia phenotypes [73]. Furthermore, compound heterozygosity for Pitx2 and Tbx1 results in alignment defects including DORV and pulmonary stenosis, further indicating how SHF deployment and laterality pathways intersect during OFT morphogenesis [74].
Multiple mechanisms intervene in OFT remodeling and impact on alignment. Rotation of the OFT myocardial wall is accompanied by spiraling of the underlying endocardial cushions that contribute to aortic and pulmonary valve leaflets. OFT cushions contain mesenchyme derived from endocardial EMT in the proximal OFT and neural crest cell influx in the distal OFT; SHF cells may also directly contribute to cushion mesenchyme [3]. Fusion of endocardial cushions plays an essential role in division of the OFT into the ascending aorta and pulmonary trunk. Defects in EMT are thought also to lead to alignment defects through disruption of cushion growth. Diverse signaling pathways are involved in cushion EMT and valve morphogenesis, including bone morphogenetic protein (BMP), transforming growth factor beta (TGFβ), and Notch signaling pathways [75]. OFT cushion defects are observed in TGFβ2 mutant mice characterized by DORV and TGA phenotypes [76]. Hemodynamics is also an important contributor to cushion remodeling: altered blood flow patterns lead to alignment defects including DORV, OA, and TGA [77]. Flow also impacts on aortic arch remodeling downstream of Pitx2 function in the OFT, suggesting that OFT rotation itself may be genetically determined rather than flow driven [78]. Differential growth of subaortic and subpulmonary myocardium has also been proposed to be essential for OFT alignment. Accordingly, either deficiency of subpulmonary myocardium or overgrowth of subaortic myocardium after progenitor cell addition to the heart would lead to a side-by-side configuration of the ascending aorta and pulmonary trunk [7, 79]. Analysis of genes differentially expressed in subaortic versus subpulmonary myocardium may generate new insights into the remodeling processes [10]. Independently of OFT rotation, a leftward shift of the OFT has been proposed to play an important, although poorly understood, role in aligning the ascending aorta with the left ventricle [80, 81]. The absence of such a leftward shift would distinguish DORV from TGA in the context of a lack of OFT rotation. Myocardial cell shape defects have also been proposed to potentially contribute to alignment defects [46]. For example, altered cell polarity in the remodeling OFT has been observed in Wnt11 null embryos, and mutation of the gene encoding non-muscle myosin IIB results in an OA phenotype associated with myocardial hypertrophy [51, 82]. Finally, programmed cell death also plays a major role in the remodeling step, in particular at the base of the ascending aorta, where patterned apoptosis is required to align the aorta with the left ventricle [83]. Blocking cell death in chick embryos using caspase inhibitors or by expression of an inhibitor of apoptosis results in DORV (TGA type) and TGA phenotypes [84].
The generation and remodeling of OFT myocardium are thus both essential for correct ventriculoarterial alignment. It is important to point out that the range of OFT phenotypes observed in mouse models and human CHD patients highlights the importance of genetic modifiers of mutant phenotypes. In addition, non-genetic factors, including teratogens, maternal diabetes or illness, epigenetic factors, and even stochastic events, can impact on either or both steps of OFT elongation and remodeling and result in alignment defects such as DORV and TOF [85].
Conclusion
In conclusion, DORV and TOF are part of a disease spectrum of OFT alignment defects arising through multiple upstream causes, reflected in the diversity of cell types, genes, and embryological processes involved in OFT extension and remodeling. While OFT alignment is distinct from neural crest-driven OFT septation, the two processes are coincident and together ensure the separation of systemic and pulmonary circulatory systems at birth. The mechanistic complexity underlying these processes is reflected in the range of associated CHDs and genetic heterogeneity observed in human patients.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


