Class
Genes
Transcription factor
Cited2, Eya1, Foxc1, Foxc2, Gata3, Gata6, Gbx2, Hand2, Pbx1, Prdm1, Prdm3, Prrx1;Prrx2, Tbx1, Tcfap2a, Zic3
Signalling molecule
Notch, Rara; Rarb, Scrib, Sema3c
Growth factor
Fgf8, Tgfb2, Vegfa
Protein binding
Bmp4, Chrd, Crkl, Edn1, Ednra, Flna, Ltbp1L, Vangl2
Receptor
Acvr1, Bmpr2, Folr1, Pdgfra, Plxnd1, Tgfbr2
Enzyme
Aldh1a2, Chd7, Ece1
42.3 T-Box 1
The gene thought to be responsible for the cardiovascular defects seen in 22q11 deletion syndrome (22q11DS, also known as DiGeorge, velocardiofacial, or Shprintzen syndrome) is T-box 1 (TBX1) [6–8]. Although clinically rare, 50 % of all cases of IAA occur in this syndrome [9]. Transgenic mice homozygous null for Tbx1 die in the perinatal period with common arterial trunk (CAT). However, mice lacking one copy of Tbx1 (i.e. heterozygous, Tbx1 +/− ) are generally healthy and fertile, but, importantly, a proportion do display fourth PAA defects such as IAA (which is lethal) and A-RSA (which may be tolerated) (Fig. 42.1). This heterozygous phenotype makes the Tbx1 +/− mouse a relatively good clinical model for 22q11DS, and transgenic embryos have been produced to investigate the tissue-specific requirements in PAA morphogenesis in the Tbx1 heterozygous state. Hypoplasia of the fourth PAA was achieved by removing Tbx1 from the second heart field, mesoderm, ectoderm and endoderm using forkhead box G1/Cre-recombinase (Foxg1Cre) transgenic mice [11]. As conditional deletion from the mesoderm using mesoderm posterior 1 homolog (Mesp1)-Cre transgenic mice does not result in PAA defects, this seems to exclude the role of Tbx1 in the mesoderm for fourth PAA morphogenesis [11]. Additional evidence for this is provided from pharyngeal epithelial deletion using fibroblast growth factor (Fgf) 15-Cre and activating enhancer binding protein 2 α (AP-2α)-IRESCre, a targeting cassette containing the Cre-recombinase coding sequence and an internal ribosome entry site (IRES), which also results in fourth PAA defects [11].
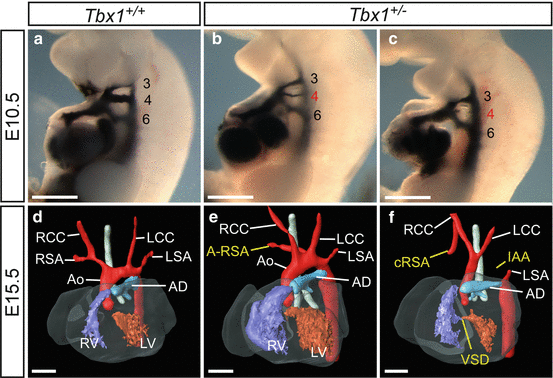
Fig. 42.1
Cardiovascular defects in Tbx1-heterozygous (Tbx1 +/− ) mouse embryos and foetuses. (a–c) Injection of India ink into the heart of E10.5 embryos. (a) In the wild-type embryo (Tbx1 +/+ ), three bilaterally symmetrical pharyngeal arch arteries (PAA) are present (numbered 3, 4 and 6) which are fully patent to ink. (b, c) In Tbx1 +/− embryos, the fourth pharyngeal arch arteries (PAA) may either be hypoplastic (b) or non-patent to ink (c) revealing that this PAA has failed to form properly. (d–f) 3-D reconstructions from MRI datasets [10] of E15.5 foetuses. (d) By E15.5, the heart and its associated great vessels have developed into the mature configuration. (e, f) The hearts of Tbx1 +/− foetuses present with great artery defects related to the failure of the fourth PAA to form correctly: A-RSA (e) and IAA (f) which, in this case, is accompanied by cervical origin of the RSA (cRSA) and a VSD. Abbreviations: Ao aorta, AD arterial duct, IAA interrupted aortic arch, LCC left common carotid; LSA left subclavian artery, LV left ventricle, RCC right common carotid, RSA right subclavian artery, RV right ventricle. Scale, 500 μm
In the zebrafish, mutant alleles of tbx1 have been generated through the DNA damaging agent ENU (N-ethyl-N-nitrosourea) [12, 13]. The two separately identified alleles cause a complete loss of function of tbx1. These tbx1 mutant zebrafish have small otic vesicles and show defects in the segmentation of the posterior pharyngeal arches. Whereas in mice or humans, the PAAs become asymmetrically remodelled, in zebrafish, the four posterior PAAs persist into adulthood and only the two anterior PAAs become remodelled [14]. In all tbx1-mutant larvae, however, the PAAs are highly reduced in number, with only one or two PAAs formed which are capable of carrying blood. Similarly to the mouse model of Tbx1 loss, the neural crest defects are secondary to endodermal defects [12]. A genetic interaction between tbx1 and vegfa has been demonstrated in the zebrafish through co-injection of morpholinos to knock down each gene simultaneously [15].
Direct targets of Tbx1 have been identified, either through microarray analysis or genetic interaction studies in vivo. For example, microarray analyses comparing Tbx1 null embryo tissue with wild-type controls have generated many potential downstream targets of Tbx1 [16, 17], although not all will directly affect PAA development. Studies in vivo have shown that Tbx1 is in epistasis, or able to genetically interact, with a number of other genes and pathways, including a bone morphogenetic protein (Bmp)-Smad pathway [18], chromodomain helicase DNA-binding protein 7 (Chd7) [19], V-Crk avian sarcoma virus CT10 oncogene homolog-like (Crkl) [20], Fgf8 [21], Fgf-Six homeobox 1 (Six1) signalling [22], gastrulation brain homeobox 2 (Gbx2) [23], hes family bHLH transcription factor 1 (Hes1) [24], paired-like homeodomain 2 (Pitx2) [25], PR domain containing 1 (Prdm1) [26], SMAD family member (Smad) 7 [27] and wingless-type MMTV integration site family, member 5A (Wnt5a) signalling [28].
42.4 Fibroblast Growth Factor 8
As mentioned above, Fgf8 is in epistasis with Tbx1. Fgf8 is an important signalling factor in PAA development, and the Fgf8 gene is expressed in the ectoderm and endoderm of the pharyngeal arch region. Mice null for Fgf8 die early in embryogenesis [29], but mice expressing a Fgf8 hypomorphic allele present with IAA, RAA (right aortic arch) and A-RSA, as well as other intracardiac and outflow tract defects such as CAT and double-outlet right ventricle (DORV) [30, 31]. Mice doubly heterozygous for Tbx1 and Fgf8 have a higher penetrance of fourth PAA defects than Tbx1 heterozygotes, i.e. IAA-B, RAA, A-RSA and cervical origin of the aortic arch [21]. The PAA defects were attributed to the loss or hypoplasia of the fourth PAA as a result of neural crest cell disruption, suggesting that Fgf8 is essential in neural crest cell survival [30]. Interestingly, an expanded expression pattern of heart and neural crest derivatives expressed 2 (Hand2) in Fgf8 hypomorphs was observed, which could suggest a complex regulatory loop between these two genes [30].
Tissue-specific removal of Fgf8 from the pharyngeal epithelia (ectoderm and endoderm) has revealed that ectodermal expression of the gene is invaluable in PAA development [32]. Removal of Fgf8 from the pharyngeal ectoderm using AP-2α-IRESCre resulted in 95 % penetrance of fourth PAA malformations at E18.5, of which 30 % were IAA. Other defects included abnormalities of the subclavian arteries, circumflex RAA with or without a right-sided arterial duct and malformations of the coronary arteries. Overall, the levels of PAA malformation mirror that of hypomorphic mutants, suggesting that the pharyngeal ectoderm is the important expression domain of Fgf8 for fourth PAA development. Early differentiation and migration of endothelial cells were unaffected in these mutant embryos; however, these cells failed to organise correctly into vascular tubes. Additionally, removal of Fgf8 from the ectoderm and endoderm using homeobox A3 (Hoxa3)-IRESCre revealed a comparable number of PAA malformations to those seen in the ectoderm-specific deletion. Thymus and parathyroid malformations, in addition to bicuspid aortic valve (BAV), were uniquely identified in these mutants, suggesting that endodermal Fgf8 expression is key in gland and valve development. Apoptosis of neural crest cells was identified within the third pharyngeal arch mesenchyme and that of the forming fourth and sixth pharyngeal arches. It appears that the ectodermal expression of Fgf8 is crucial for the formation of the fourth pharyngeal arch; however, the defects observed in Fgf8;Hoxa3-IRESCre mutants are perhaps the result of a synergistic effect of Fgf8 removal from both the ectoderm and endoderm simultaneously. A signalling pathway between the neural crest-derived mesenchyme and endothelial cells is apparently required for correct formation of the PAA, at a time prior to differentiation of neural crest cells into vascular smooth muscle cells [32].
42.5 Gastrulation Brain Homeobox 2
Another gene that has been linked to Tbx1, as a potential downstream target, is the transcription factor gastrulation brain homeobox 2 (Gbx2) [16]. Mice null for Gbx2 present with a range of cardiovascular malformations including IAA and A-RSA, as well as a small number of outflow tract defects, such as overriding aorta and DORV [23, 33]. Reminiscent of the Tbx1-null phenotype, these malformations appear to be related to disrupted neural crest cell migration at E8.0–9.5 of mouse embryonic development. However, as Gbx2 is not expressed in neural crest cells, it was determined that Gbx2 expression within the pharyngeal epithelia is responsible for the activation of signalling pathways required in neural crest cell migration into the pharyngeal arches [23]. Conditional deletion of Gbx2 from the pharyngeal ectoderm, using AP-2α-IRESCre, resulted in a comparable level of fourth PAA defects as seen in Gbx2 −/− embryos, when examined at E10.5 by ink injection. This suggests that ectodermal expression of Gbx2 is key to the neural crest cell migration defects observed in Gbx2 −/− embryos. A fusion of the neural crest cell streams as they entered the pharyngeal arches was also observed. These defects were found to be the result of disruption of the Slit homolog (Slit)/roundabout axon guidance receptor homolog (Robo) pathway, involved in migration of the neural crest cells to the pharyngeal arches [23].
Additionally, there is evidence to support a potential in vivo genetic interaction between Gbx2 and Tbx1[23]. Doubly heterozygous mutant embryos (i.e. Tbx1 +/− ;Gbx2 +/− ) show a statistically significant increase in fourth PAA malformation at E10.5 than that observed in Tbx1-heterozygous mutant embryos (i.e. Tbx1 +/− ;Gbx2 +/+ ). In addition, higher numbers of embryos showed bilateral defects instead of unilateral defects. Furthermore, reduced expression of Slit2 was also observed in Tbx1 +/− mutant embryos, and those with a pharyngeal ectoderm-specific deletion, to a greater extent than that seen in Gbx2 mutants. This suggests that Tbx1 may also be capable of regulating Slit2, independent of Gbx2.
A further genetic interaction has been proposed between Gbx2 and Fgf8. The expression patterns of both Gbx2 and Fgf8 overlap, suggesting that the two genes may be interlinked, although there is no change in the expression levels of Fgf8 in Gbx2 mutant embryos [33]. However, Gbx2 can be induced by ectopic FGF8 expression, and Gbx2 expression in E8.5 Fgf8 hypomorphs was found to be reduced in the region overlying the future fourth PAA. This, therefore, suggests that Gbx2 is a downstream effector of Fgf8 signalling (Fig. 42.2). This notion was confirmed by changes in the number of PAA defects in complex mutant embryos. Double heterozygous embryos (i.e. Gbx2 +/− ;Fgf8 +/− ) showed an increase in the incidence of A-RSA and RAA, whilst no defects were observed in either of the single heterozygotes. Generating embryos homozygous null for Gbx2 and heterozygous for Fgf8 (i.e. Gbx2 −/− ;Fgf8 +/− ) resulted in a significant increase in the number of PAA malformations, compared to those seen in Gbx2 −/− embryos. It is proposed that Tbx1 is upstream of Fgf8, which then activates Gbx2 and results in correct PAA development. It is also postulated that Fgf8 is capable of acting independently, through a parallel pathway which controls neural crest cell survival, in forming the PAA, as neural crest cell apoptosis is observed in Fgf8 mutants, but not in Gbx2-null embryos [32, 33].
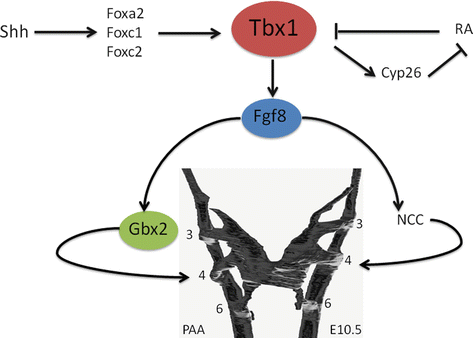
Fig. 42.2
Schematic model of the genes discussed in this chapter and their involvement in regulating Tbx1 and the development of the fourth pharyngeal arch arteries (PAA). Abbreviations: Cyp26 cytochrome P450, family 26 protein, Fgf8 fibroblast growth factor 8, Fox forkhead box protein, Gbx2 gastrulation brain homeobox protein 2, NCC neural crest cells, RA right atrium, Shh sonic hedgehog, Tbx1 T-box protein 1 (Figure adapted from Refs. [33, 34])
42.6 Transforming Growth Factor-β Signalling
Transforming growth factor (TGF)-β signalling is crucial for regulating basic cell responses, including proliferation, migration and apoptosis and has been shown to be important for embryonic development in mouse models. The TGFβ signalling pathway is initiated by the binding of TGFβ ligand to the cell surface TGFβ type II receptor (TGFBR2), which activates the TGFβ type I receptor, TGFBR1 (also known as ALK5), and phosphorylates SMAD2 and SMAD3 proteins, allowing association with SMAD4 and translocation to the nucleus to regulate downstream gene expression.
Transgenic mice null for the TGFβ2 ligand (Tgfb2) die perinatally with cardiovascular defects, including IAA and A-RSA, as well as VSD, DORV and CAT [35, 36]. Aberrant apoptosis within the fourth PAA was observed, without any defects in neural crest cell migration, which may contribute to the IAA phenotype as well as causing problems with its innervation [36, 37]. The TGFβ receptor Tgfbr2 is essential for embryogenesis, as mice lacking this gene die around mid-gestation [38, 39]. The conditional deletion of Tgfbr2 from the neural crest results in IAA [40]. Disruption of the Ltbp1 gene, which encodes for the long form of latent TGF-β-binding protein 1 and is required for correct storage of TGFβ ligands in the extracellular matrix, also results in embryos with IAA [41].
42.7 Coarctation
Coarctation of the aorta is defined as a narrowing of a section of the aorta that will restrict the flow of blood from the heart. This defect has been recognised in mouse models with heart and great vessel defects, but rarely as an isolated phenotype. It is much more commonly seen within the context of complex cardiovascular malformations.
Aortic coarctation is present within the spectrum of cardiovascular phenotypes seen in 22q11DS patients, and coarctation has also been seen in mouse models genetically connected to Tbx1. The expression of Tbx1 is induced and sustained by Sonic hedgehog (Shh) in the pharyngeal endoderm [43]. However, the forkhead transcription factors Foxa2, Foxc1 and Foxc2 have been shown to recognise and bind to cis-regulatory elements in the Tbx1 promoter and to directly promote the expression of Tbx1 [44, 45]. It is proposed that Tbx1 is a direct transcriptional target of Fox proteins, which play an intermediary role in the regulation of Tbx1 by Shh (Fig. 42.2). Mice null for Foxc1 have coarctation and aortic and pulmonary valve dysplasia, as well as IAA-B and patent arterial duct [46], and mice null for Foxc2 have IAA-A and IAA-B [46, 47]. The different descriptions for IAA reflect the location of the interruption: IAA type A is found when the segment of the aortic arch distal to the left subclavian artery is interrupted, IAA type B occurs when the segment of the aortic arch between the left common carotid and left subclavian artery is missing, and type C is when the aortic arch between the right and left common carotid arteries is absent. Mice doubly heterozygous for Foxc1 and Foxc2 (i.e. Foxc1 +/− ;Foxc2 +/− ) also present with coarctation and aortic and pulmonary valve dysplasia and also have IAA-A and IAA-B [46], whereas individual heterozygotes do not.
Folr1 is a folate-binding protein gene, and mice null for Folr1 die at mid-embryogenesis from extensive cell death. However, folic acid supplementation of pregnant females greatly reduces this early embryonic lethality and allows for Folr1-null mice to be born and bred [48]. A small proportion of these Folr1-null foetuses had coarctation of the aortic arch or IAA, as well as a right-sided aortic arch and A-RSA. In these foetuses, coarctation was found proximal to the arterial duct.
Coarctation has also been described in transgenic mice doubly homozygous for the retinoic acid receptors alpha and beta (Rara −/− ; Rarb −/− ), and additionally these mice present with CAT, right-sided aortic arch and A-RSA [49]. Also, mice null for Vangl2, a membrane protein involved in the regulation of planar cell polarity, and the gene mutated in the loop-tail mouse, die from complex cardiovascular defects that include preductal coarctation as well as IAA, DORV, VSD and right-sided aortic arch [50].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


