Fig. 54.1
(a) Location of the ring of musculature of the primary heart tube delineated by reaction to an antibody to the nodose ganglion of the chick. This is the myocardium of the so-called primary ring. (AV atrioventricular, GIN2 antigen to chick nodose ganglion). (b) The first step in remoulding of the primary ring occurs concomitant with appearance of the primary ventricular septum. As the apical parts of the ventricle balloon form the primary tube, the inferior part of the primary foramen deepens to form the right ventricular inlet component between the right atrium and the dorsal part of the developing right ventricle. At the same time, the cushions within the atrioventricular canal become draped across the inferior part of the forming ventricular septum (Adapted from [13])
In the first phase, the parietal wall of the right atrium will come into continuity with the parietal wall of the right ventricle through remodelling and posteroinferior expansion of the myocardium at the right junction. Concomitant with the development of the right atrioventricular connection, a myocardial ridge forms at the boundary between the AV canal and the embryonic right ventricle. This myocardial funnel develops to become the “tricuspid gully” [12], which at this stage does not have any leaflets of the tricuspid valve yet. It grows to become a myocardial gully that funnels atrial blood beneath the lesser curvature of the initial heart tube towards the middle of the right ventricle. Fenestrations in the floor of the gully create an additional inferior opening in the funnel, with the primary orifice of the developing tricuspid valve running parallel to the developing ventricular septum [13].
In the second phase, epicardially derived cells, myocardium and endocardial cushions all make contributions to the leaflets of the tricuspid valve [12, 13, 16]. With the appearance of the endocardial cushion, the lumen between the trabecular and compact layers of myocardium expand, leading to the formation of the anterosuperior and inferior leaflets during the seventh week of gestation. Delamination of the septal leaflet from the developing ventricular septum occurs much later during the twelfth week. The myocardial contributions to the septal leaflet come from both the myocardial gully and the ventricular septum.
In parallel to this process, the insulation between the muscle masses of atrium and ventricle is established by the fusion of the tissues of the atrioventricular sulcus (located at the epicardial side of the junctional myocardium) with those of the atrioventricular cushions (located at the endocardial side of the junctional myocardium). This fusion leads to the formation of fibrous insulating tissues at approximately 7 weeks of development in the anteromedial portion of the right atrioventricular junction and is largely completed around the 12th week of development. In the normal adult heart, the only remaining myocardial continuity between atrial and ventricular myocardium is the atrioventricular axis of conduction [15].
Molecular pathways involved in the pathogenesis of Ebstein anomaly are likely to impact the cellular and morphological events of tricuspid valve formation at very early stages. Several signalling and transcription cascades are therefore prime candidates based on their spatiotemporal expression patterns or known functions in heart morphogenesis.
Gain- and loss-of-function studies, tissue explant studies and expression patterns point towards an important role of BMP (bone morphogenetic protein) signalling in endothelial-mesenchymal transformation of endocardial cushions. BMP2 is strongly expressed in AV cushions, whereas the major BMP receptors, including BmprII, Alk2 (Acvr1, activin A receptor, type 1) and Alk3 (Acvr3) are widely expressed in the entire heart, with no clear spatial restriction [17, 18]. Using a GATA (GATA binding protein) 6-Cre driver mouse, Gaussin et al. conditionally deleted Alk3 in the developing AV canal [19]. Interestingly, lineage analysis showed a clear contribution of AV canal cardiac myocytes to the future tricuspid mural and posterior leaflets, but not to the tricuspid septal leaflet. The mitral septal leaflet and the atrial border of the annulus fibrosus were also stained. When Alk3 was deleted in these cells, defects were seen in the same leaflets, i.e., the tricuspid mural leaflet and mitral septal leaflet were longer, the tricuspid posterior leaflet was displaced and adherent to the ventricular wall and the annulus fibrosus was disrupted resulting in ventricular preexcitation. These defects provide in fact some overlap with Ebstein anomaly and demonstrate that signalling by Alk3, the type 1a receptor for bone morphogenetic proteins, in the myocardium of the AV canal is required for the development of both the AV valves and annulus fibrosus, but they do not replicate the hallmark of the condition, namely, the apical displacement and non-delamination of the septal leaflet of the tricuspid valve. Conceivably, expression of Cre in the GATA6-Cre driver mouse may become progressively restricted in primary myocardium and thus insufficient to label all AV canal myocardial cells.
However, in extrapolation of these results, it can be presumed that key processes possibly involved in Ebstein anomaly are at least in part mediated through BMP signalling in the developing atrioventricular myocardium, including the processes leading to proper electric insulation of ventricles and atria [19]. Within the same morphological processes and molecular pathway, it was observed that positioning of the endocardial cushions in the ventricular lumen and formation of early trabeculae are part of one remodelling process which is under control of TGF-β2 (transforming growth factor, beta 2) [20]. In the Tgf-beta2-KO mouse, remodelling of the early myocardium is disturbed as indicated by defective trabeculae formation, persistence of valvular myocardium, disturbed myocardial phenotypes and differential defects at left and right side of the AV canal [20]. Bmp2 and Tgf-beta2 themselves are under genetic control of Tbx (T-box) 20, disruption of which leads to defective AV canal myocardial and valve development [21].
Epicardially derived cells (EPDC) are known to contribute to the developing fibroblast population in the heart [22, 23]. EPDCs contribute to the development of the AV canal and the annulus fibrosus and populate the ventricular myocardial wall and interventricular septum [24, 25]. While early development of the myocardial compact layer is independent of EPDCs, the subsequent increase in wall thickness is impaired when epicardial development is disrupted [26, 27]. Using the mWt1/IRES/GFP-Cre mouse to trace the fate of EPDCs from embryonic day (ED) 10 until birth, Wessels et al. found that there is a dramatic disparity of the contribution of epicardial- versus endocardial-derived cells to the different leaflets of the right-sided AV valve: the parietal AV leaflets have a very strong epicardial contribution, whereas the septal leaflet of the tricuspid valve (and all leaflets derived from the major cushions) hardly have any epicardial contribution [16]. The differential contribution of EPDCs and other cell sources to the various leaflets of the atrioventricular valves, as well as their roles in myocardial growth and maturation, thus helps to understand how individual components of this region of the heart may be differentially affected in Ebstein anomaly. A corresponding cell population that heavily populates the septal leaflet of the tricuspid valve and also participates in the remodelling of the “tricuspid gully” [12], however, remains elusive to date. Equally unknown are the precise molecular factors that drive the apoptosis of the myocardial layer in the developing valves and their subsequent excavation and delamination [28, 29].
54.3 Mouse Models of Ebstein Anomaly
Although several mouse models exist that replicate some aspects of Ebstein anomaly, no single-gene model exhibiting all its features has been reported to date. A model of tricuspid atresia was reported in Fog2 (friend of GATA 2 or ZFPM2) knockout mice, most likely secondary to impaired attenuation of endothelial-to-mesenchymal transition within the developing cushions [30, 31]. The recently reported Nkx2-5 (NK2 homeobox 5) p.Arg25Gly knockin allele, which mimics a mutation found in humans, is the most faithful murine Ebstein model to date [32]. The human NKX2-5 p.Arg52Gly mutation destabilises the homeodomain of this transcription factor, reduces DNA binding as well as homo- and heterodimerisation with GATA4 and TBX5 [33]. In the 129Sv background, this knockin leads to a 100 % penetrance of CHD, in particular LVNC (100 %), ventricular septal defects (82 %), atrioventricular septal defects (18 %) and tricuspid valve anomalies (47 %). Incomplete delamination was seen in all mice with tricuspid valve anomalies, with all hallmarks of Ebstein anomaly in 8.5 % of mice. Detailed molecular assessment and timelines of maldevelopment of the tricuspid valve have not yet been reported in this mouse. In keeping with other studies [34, 35], penetrance of different phenotypic aspects of those mice was highly dependent on the background. After one cross into the C57B6 background, attenuation of most defects was noted, with the exception of LVNC which still had a very penetrance [32]. As seen in other mouse mutants, these important observations suggest that the morphogenetic processes governing normal and abnormal tricuspid formation are likely to be under the control of a cooperative gene network [36].
54.4 Canine Tricuspid Valve Malformation As a Model for Human Ebstein Anomaly
Dogs have long been popular models for cardiac physiology studies because canine heart size approximates the human heart. The advantages of the canine as a potential model of genetic heart disease include short generation time and multiple offspring in one litter. Selective breeding strategies increase the likelihood of transmission of desirable traits, but also lead to an excess of genetic disorders [37]. Individual dogs may have more than 100 offspring and exert a strong influence on the genetic outfit of a particular breed (“popular sire effect”) [38]. Therefore, founder effects play an important role in modern dog breeds, even in the absence of known common ancestors [39, 40]. As an example, there is statistical genetic and molecular evidence for cardiovascular traits as diverse as subaortic stenosis and cardiomyopathy that strong genetic factors shape the phenotype, albeit this can occur in complex fashion [41–43].
Canine tricuspid valve malformation (CTVM) is characterised by an apical displacement of the attachment of the tricuspid leaflets and adherence of the septal leaflet to the interventricular septum (Fig. 54.2). Other features include thickening of the valve leaflets, redundancy of the parietal leaflet and presence of a large, fused papillary muscle rather than normal small, discrete muscles. Additional congenital heart defects in affected dogs have been described, including mitral valve abnormality [45–49]. CTVM has been most frequently identified in Labrador Retrievers, but it has been observed in other breeds as well [47, 50–53]. As in humans, the clinical picture varies, with severity in affected dogs ranging from asymptomatic to premature death secondary to right heart failure [48, 50, 51]. In conclusion, CTVM mimics the morphological and clinical picture of Ebstein anomaly.
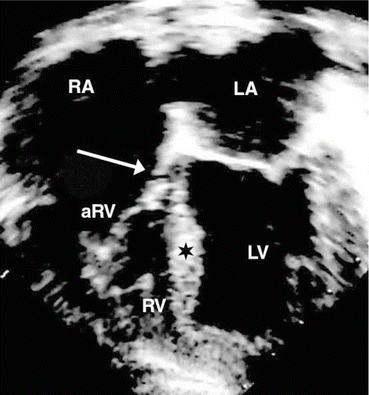
Fig. 54.2
Apical four-chamber echocardiogram in canine tricuspid valve malformation. Arrowhead downward displacement and abnormal adherence of the septal leaflet of the tricuspid valve [44]. RA right atrium, LA left atrium, aRV atrialised right ventricle, RV right ventricle, LV left ventricle, ★ interventricular septum
The availability of larger pedigrees and ascending genealogies from breeding documents has facilitated the genetic analysis of CTVM. All three available studies show clear evidence of heritability of CTVM, two of them in favour of a recessive mode of transmission [52, 53] and one of them in favour of an autosomal dominant transmission with reduced penetrance [44]. In this latter study, a total of 140 tested polymorphic markers excluded ~60 % of the genome before suggestive linkage to canine chromosome 9 (CFA9) was detected at REN75M10 with a two-point LOD score of 1.56. Subsequently, fine mapping with adjacent and linkage analysis yielded a maximum multipoint LOD score for the three most informative markers within the critical interval of 3.33, indicating odds of greater than 2000:1 that CTVM is linked to a locus on CFA9 (Fig. 54.3) [44]. Subsequent comparative mapping showed that this interval is homologous to a gene-rich region on human chromosome 17q, but a disease-contributing variant could not yet be isolated (Fig. 54.4) [54, 55].
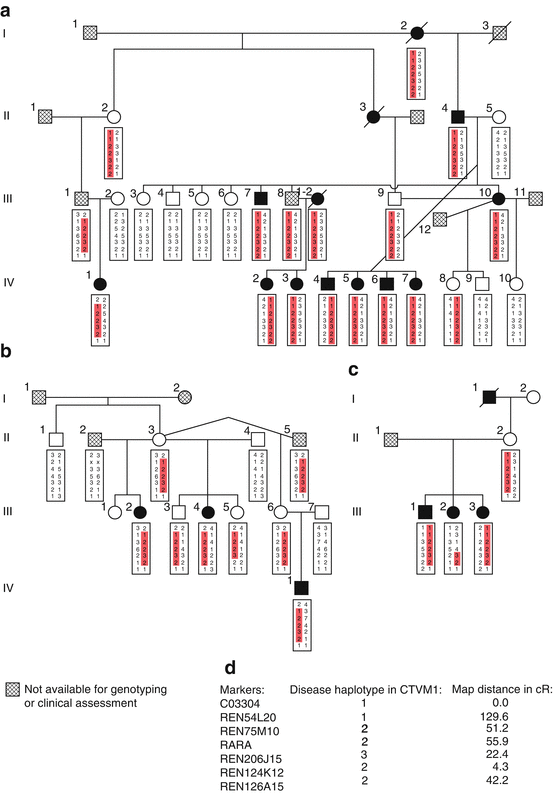
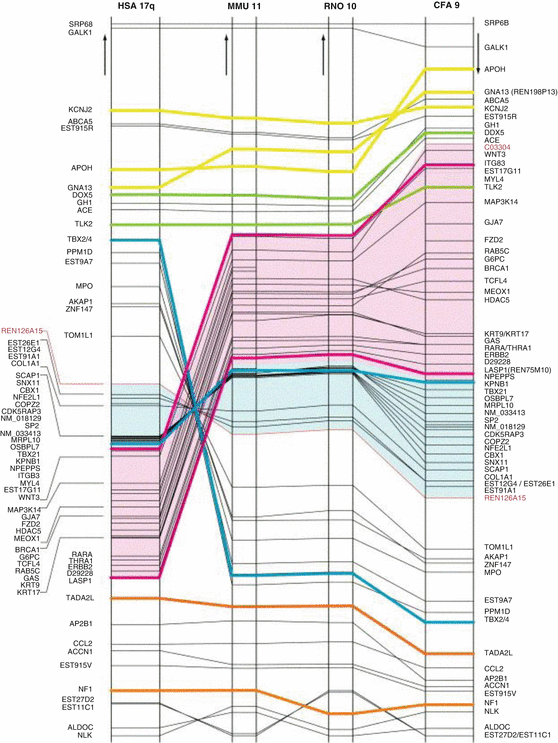
Fig. 54.3
(a) kindred CTVM1, (b) kindred CTVM2, (c) kindred CTVM3, (d) legend, (e) genotyped markers and map distances (cR). All affected dogs in kindreds CTVM2 and CTVM3 also fulfilled the diagnostic criteria for canine tricuspid valve malformation (CTVM). None of the dogs in the series reported here showed any other intra- or extracardiac malformation [44]
Fig. 54.4
Horizontal axis: human chromosome 17q (HSA17q) and its syntenic regions on mouse chromosome 11 (MMU11), rat chromosome 10 (RNO10) and dog chromosome 9 (CFA9). Vertical axis: Chromosomal positions of genes, with largest distances in individual species equalised to each other. Positions are drawn to scale. Arrows denote directionality from centromere to telomere. The recombinant markers for the ctvm interval, C03304 and REN126A15, are labelled by red lettering in the right-hand column. Yellow, green, blue, purple and orange lines facilitate the identification of genes involved in important rearrangement events. Pink and blue overlays identify human chromosomal segments that undergo macroreversals in a multiple genome rearrangement scenario. Brackets group genes in densely mapped areas in respective order [54]
Several observations are noteworthy: a single locus seems to control the predisposition in the Dogue de Bordeaux population in Israel towards both CTVM and subaortic stenosis [52]. It is unknown whether this locus is identical to the Labrador CTVM locus on CFA 9. In Labradors, two independent studies estimated heritability at 0.71 and penetrance at 68 % [44, 53]. In Keeshonds, conotruncal defects (CTD) are oligogenic, with the strongest support for a locus on CFA9 (LOD 3.7), fully overlapping with the CTVM locus mapped in Labradors [56]. Two other loci were mapped in this study to CFA2 and CFA15 (LOD scores of 2.71 and 3.03), and a two-locus analysis suggested that CTD-predisposing alleles of these three loci are necessary, at least in pairs, to produce CTD [56]. It is conceivable that breed-specific background effects in the Labrador, Keeshond and Dogue de Bordeaux dogs influence both penetrance and expressivity of CTVM, reminiscent of observations both in humans and mouse.
Conclusion
In Ebstein’s malformation, the attached margin of the septal and inferior leaflets of the tricuspid valve are displaced apically, but never beyond the junction of the ventricular inlet with the apical trabecular component [12]. This junction corresponds to the anterior limit of the myocardial gully, the expansion of which corresponds to the atrialisation of the right ventricle. The septal leaflet is not delaminated properly from the ventricular septum, with a large offset between the hinge points of the tricuspid valve and the anterior leaflet of the mitral valve. In more severe cases of Ebstein anomaly, dysplasia of the septal leaflet can be so severe that only a small orifice from the right atrium to the right ventricle persists [57–59]. It has been postulated that these features are in fact typical for the state of the normal tricuspid valve at 8 weeks of development [12].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


