Fig. 39.1
Laterality in mutant mouse models. Ap1b1 m/m mutants can exhibit a spectrum of laterality phenotypes spanning from normal situs solitus (a), to complete mirror symmetric situs inversus (b), or randomized visceral organ situs referred to as heterotaxy (c). In situs solitus, the heart apex (arrow) points to the left (levocardia), four lung lobes are on the right and one on the left, stomach is to the left, and the dominant liver lobe is on the right. With situs inversus, complete mirror reversal of organ situs is observed with both the heart and stomach on the right, while heterotaxy involves randomized visceral organ situs such as dextrocardia with levogastria shown in (c). The heterotaxy mutant in c exhibits complex CHD with atrioventricular septal defect (AVSD) (d), ventricular septal defect (VSD) (e), duplicated inferior vena cava (IVC) (f) and left pulmonary isomerism with bilateral single lung lobes (g). Ao aorta, L1–5 lung lobes 1–5, Lv1–3 live lobes 1–3, mLA morphologic left atrium, mLV morphologic left ventricle, mRA morphologic right atrium, mRV morphologic right ventricle, PA pulmonary artery, Stm stomach (From Li et al. [3])
Patients with heterotaxy usually have complex structural heart defects with anomalous left-right atrioventricular and ventriculoarterial connections that disrupts systemic vs. pulmonary circulation. This causes high morbidity and mortality, as it prevents efficient oxygenation of blood from air in the lung. Left-right patterning also can disrupt development of the biliary duct causing biliary atresia [5]. In the absence of a functional bile duct, bile backs into the liver and causes liver cirrhosis, one of the most common causes for pediatric liver transplantation. Left-right patterning defects also can cause malrotation of the intestine, with bowel obstruction arising with formation of a volvulus where a loop of the intestine is twisted along the mesentery, a life-threatening condition that requires surgical remediation [6]. Together, these findings show disruptions in left-right patterning have important clinical ramifications, and only with more mechanistic insights into the developmental regulation of left-right patterning can we hope to improve the management of heterotaxy patient care.
39.2 Nodal and TGFβ Signaling in the Embryonic Node
Studies using different model organisms have identified multiple evolutionarily conserved signaling pathways contributing to the specification of left-right asymmetry. In many vertebrate embryos, the specification of left-right patterning involves a specialized tissue referred to as the L-R organizer, also known as the embryonic node in mice (Fig. 39.2), Kupffer’s vesicle in zebrafish, the gastrocoel roof plate (GRP) in Xenopus, or Hensen’s node in the chick. The asymmetric expression of genes in the embryonic node provides some of the earliest molecular signatures showing the evolving specification of visceral organ asymmetry (Fig. 39.3). One such gene, Nodal, a transforming growth factor beta (TGFβ) family member, is initially expressed bilaterally symmetric in the perinodal crown cells, but as development progresses, expression becomes elevated on the left side of the node (Fig. 39.3).
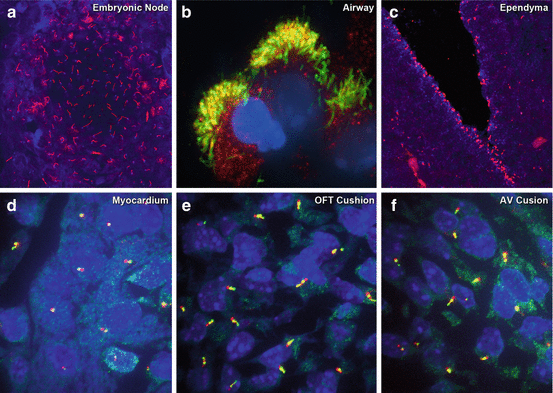
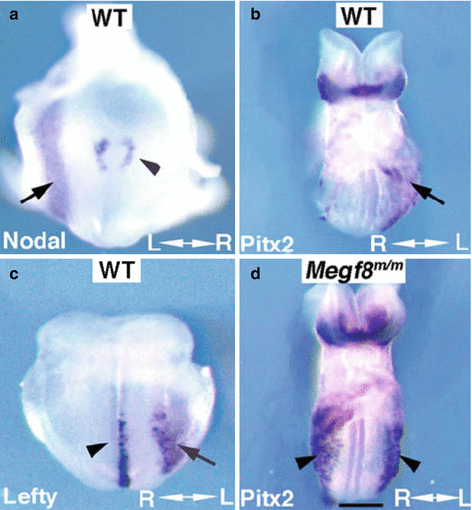
Fig. 39.2
Motile and primary cilia in different tissues of the mouse embryo. (a) Immunostaining with antibodies to acetylated tubulin (red) and γ-tubulin (blue) was used to visualize motile cilia in the E7.75 mouse embryonic node. (b–f) Immunostaining with antibodies to acetylated tubulin and IFT88 visualized cilia in the newborn mouse tracheal airway epithelia (b), in E12.5 brain ependyma (c), and primary cilia in the myocardium (d), outflow tract cushion (e), and atrioventricular cushion (f) of the E12.5 mouse embryonic heart
Fig. 39.3
Left-sided expression of left-determinant genes in mouse embryos. In situ hybridization analysis showed (a, b) left-sided expression of Nodal (a), Lefty (b), and Pitx2 (c) in wild-type embryos. Note Nodal expression is reduced in the right (see arrowhead) vs. the opposing left side of the node (a). Lefty panel (b) shows expression Lefty1 expression (arrowhead) on the left side of the floor plate, while Lefty2 expression (arrow) is seen in the left LPM. PItx2 expression was observed in the left LPM and bilaterally in the head fold (c). In contrast, embryo with Megf8 m/m mutation known to cause laterality defects (d) shows bilateral Pitx2 expression in the right and left LPM (From Zhang et al. [7])
This left dominant expression of Nodal is propagated to the left lateral plate mesoderm (LPM) (Fig. 39.3), causing the induction of Lefty2 (left-right determination factor 2), another TGFβ family member, and also the transcription factor Pitx2, the master regulator of left-sided tissue morphogenesis. Lefty2 is a diffusible competitive inhibitor of Nodal, and being monomeric, it diffuses more rapidly than the functionally active dimeric Nodal, allowing it to repress Nodal signaling in the right LPM. Additionally, expression of the related family member, Lefty 1, along the axial midline, provides a barrier function that further ensures Nodal signaling is restricted to the embryo’s right side. Ultimately, it is the Nodal-activated expression of Pitx2 (paired-like homeodomain 2) on the embryo’s left side that drives left-sided visceral organ morphogenesis.
39.3 Complex Regulation of Nodal
Nodal plays an important role not only in left-right patterning but also in specification of the anterior-posterior axis, indicating the integration of left-right patterning with anterior-posterior axis specification. The winged-helix transcription factor FoxH1 (forkhead box H1) acts upstream of Nodal (Fig. 39.4), with FoxH1-deficient mice exhibiting defects including anterior-posterior patterning defects, and failure of the node to form, phenotypes recapitulated by the loss of Nodal signaling [8]. Nodal and FoxH1 also show overlapping expression in the node [9], and expression of FoxH1 can induce Lefty2 expression asymmetrically in both the Xenopus and mouse embryos [9, 10]. Nodal expression at the node is also subject to regulation by Notch signaling [11]. This is indicated by the finding of several binding sites for RBP-J (recombination signal binding protein for immunoglobulin kappa J region), the main transcription factor mediating Notch signaling, in the Nodal enhancer. In addition, mouse mutant homozygote for the Notch ligand Dll1 or doubly mutant for Notch1/Notch2 [11] exhibit left-right patterning defects.
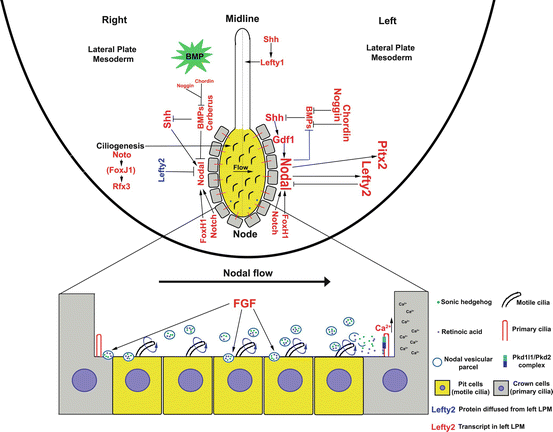
Fig. 39.4
Signaling pathways specifying left-right asymmetry. Schematic summary of cell signaling pathways contributing to the specification of laterality based on work in the mouse model. Left-right patterning requires cilia in the embryonic node. Indicated are pit cells (yellow) in the embryonic node with motile cilia driving unidirectional fluid flow. This causes mechanosensory transduction of primary cilia in the left perinodal crown cells and the elevation of calcium. A cascade of molecular signals help to propagate Nodal signaling on the embryo’s left side, defining left identity. In contrast, Nodal signaling is repressed on the embryo’s right and, together with high levels of BMP signaling, help define right identity. Unlike Lefty2, Bmp expression is bilaterally symmetric, but the green star serves to indicate high level of BMP signaling only on the right
Nodal is also subject to positive and negative regulation by other TGFβ family members, with GDF1 (growth differentiation factor 1) being an upstream positive regulator highly expressed in the node, acting to activate and propagate the Nodal signaling cascade on the embryo’s left side. Thus, Gdf1 knockout mouse embryos exhibit left-right patterning defects, such as situs inversus totalis or heterotaxy with congenital heart defects and right pulmonary isomerism [12]. This is associated with absence of Lefty1 and 2 expression and failure in Nodal propagation to the left LPM.
Nodal expression mediating left-right patterning is also subject to negative regulation by various components of the TGFβ signaling pathways. In mouse embryos, Cerberus-2 (Cerl2), also known as Dand5 (DAN domain family member 5, BMP antagonist), or Coco in the Xenopus embryo, a TGFβ antagonist, is expressed on the embryo’s right side, serving to repress Nodal expression on the right. However, beyond the 3-somite stage, Cerl2 protein shows nodal flow-dependent accumulation on the embryo’s left side, while Cerl2 transcript expression remains right-sided. This translocation of Cerl2 protein to the embryo’s left side at later stages of development may serve to shut down Nodal expression on the left as the left-right axis is established [13].
39.4 Bone Morphogenetic Protein Regulation of Nodal Signaling and Right-Sided Identity
BMP (bone morphogenetic protein) signaling is also observed to play an essential role in left-right patterning, exerting both positive and negative regulation on Nodal signaling. While BMP-4 is required for left-right patterning, its expression is actually bilaterally symmetric. This apparent discordance is explained by the higher left-sided expression of the BMP antagonists, Noggin (Nog) and Chordin (Chrd), which represses BMP signaling on the embryo’s left side, allowing left-sided Nodal propagation in the LPM [14, 15]. The essential role of BMP signaling in left-right patterning is further demonstrated by the finding that deficiency in BMP type II or type I receptors, Acvr (activin A receptor) IIb [16] and Acvr1 [17], respectively, causes left-right patterning defects. While these and other studies clearly show BMP signaling playing important roles in restricting Nodal activation to the right LPM, its right-sided activation may also help define right-sided identity.
39.5 Embryonic Node and Left-Right Patterning
The important role of Nodal signaling in patterning left-right visceral organ morphogenesis is unequivocal, but how the Nodal signaling cascade brings about the left-right asymmetric patterning of visceral organ morphogenesis is not well understood. The embryonic node has been the focal point of such studies. In the mouse embryo, it is formed at the anterior tip of the primitive streak at the future posterior side of the embryo. While it is often said that breaking of left-right symmetry occurs with asymmetric activation of nodal signaling at the embryonic node, left-right symmetry is already established earlier. Thus with formation of an anterior-posterior and dorsoventral axes in the pregastrulating mouse embryo, a left-right axis is defined by default [18]. The node subsequently forms at the anterior tip of the primitive streak, playing a critical role in the differential propagation of the Nodal signaling cascade to specify left identity. This regulation involves motile cilia that exhibit clockwise rotation creating a leftward fluid flow. In addition, there are also nonmotile primary cilia in cells at the node periphery referred to as perinodal crown cells. These have been shown to have a mechanosensory function in flow sensing and may help relay calcium signaling to propagate Nodal signaling.
39.6 Motile Cilia in Left-Right Patterning
The requirement for motile cilia in left-right patterning is suggested by the finding of laterality defects in mutants with disruption of motile cilia function. Insights first came from observations of mutations in two genes encoding kinesins, Kif3a and Kif3b [19, 20]. Kinesins are molecular motors which traffic cargo along microtubule tracks and, importantly, are required for the assembly of cilia through intraflagellar transport. These kinesin mutant mice not only failed to form cilia and showed no nodal fluid flow, approximately 50 % displayed laterality defects.
In reality, the connection between motile cilia and abnormalities of the situs has long been known clinically through observations of patients with Kartagener syndrome [21], a condition in which patients can exhibit situs inversus totalis together with sinopulmonary disease and male infertility due to motile cilia dysfunction, a condition referred to as primary ciliary dyskinesia, or PCD (see Chap. 38). While the mechanism by which motile cilia defect caused laterality defects in PCD patients was unknown at the time, the requirement for motile cilia function in left-right patterning was clearly indicated by these findings.
Corroborating these clinical findings with PCD patients were observation of the inversus viscerum (iv) mutant mice. This mutant was later shown to harbor a mutation in the dynein gene, Dnah (dynein, axonemal, heavy chain) 11, a gene now demonstrated to be a common cause of PCD [22]. In iv mutant mice, nodal cilia are formed but are rendered immotile by the Dnah11 deficiency, causing the disruption in left-right patterning. Such motile cilia defects underlie the sinopulmonary disease associated with PCD. Thus Dnah11 is required for motile cilia function mediating mucus clearance in the airway and in left-right patterning in the embryonic node.
Since these early studies, many cilia-related genes have now been identified to cause PCD and laterality defects, including genes encoding other dyneins such as DNAH5 [23, 24] and DNAI1 [25, 26], as well as many other cilia-related genes [3]. These studies established a clear link between motile cilia defects and defects in left-right patterning. Two transcription factors, FoxJ1 (forkhead box J1) and Rfx3 (regulatory factor X, 3, influences HLA class II expression), are observed to play important roles in ciliogenesis at the node, with Foxj1 also referred to as a master regulator of ciliogenesis [27]. While Foxj1 knockout mice and morpholino knockdown of foxj1 show loss of cilia [28–30], ciliogenesis was not affected in a Foxj1 mouse mutant harboring a missense mutation in the conserved DNA binding domain [3]. Cilia were observed both in the airway and in the embryonic node of this mutant, although ciliary motion was dyskinetic in the node. These findings suggest the intriguing possibility that Foxj1 may also have a non-transcriptional role in the regulation of cilia function.
Exactly how motile cilia regulate left-right patterning is still not well understood. Currently there are two predominant models, one referred to as a “morphogen” hypothesis that suggests motile cilia play a role in transporting morphogen to the embryo’s left side to specify left identity, while a second model referred to as the “two-cilia” hypothesis proposes motile cilia generate nodal flow propagates Nodal signaling via mechanosensory transduction of primary cilia in the perinodal crown cells. Below we briefly discuss evidence in support of these two models.
39.7 The “Morphogen” Hypothesis
The “morphogen” hypothesis proposes that cilia-generated flow in the node provides leftward transport of a secreted morphogen that establishes left identity [31]. This model builds on the observation of left ward transport of membrane vesicles up to 5 μm in size termed nodal vesicular parcels (NVPs) in the node. These NVPs, released from microvilli, are swept across the node by cilia-generated flow. Upon coming into contact with the rotating cilia, the NVPs undergo fragmentation, releasing smaller particles that accumulate along the left wall of the node. Significantly, these NVPs contain sonic hedgehog (SHH) and retinoic acid (RA), both known to play essential roles in left-right patterning (Fig. 39.4) [32, 33]. This left-sided concentration of morphogens from the NVPs is further proposed to trigger left-sided signal transduction mediated by sensory cilia in the perinodal crown cells, resulting in left-sided elevation of Ca2+, and the propagation of Nodal expression, and asymmetric organ patterning.
Consistent with this NVP model, Kif3a-deficient embryos with no nodal cilia were observed to have significantly more NVPs [31]. Similarly, in Dnah5 mutant embryos with paralyzed nodal cilia, we observed a similar accumulation of large vesicles, suggesting NVPs may fail to undergo fragmentation in Dnah5 mutant embryos with paralyzed cilia (Fig. 39.5) [23]. Also of interest is our recent recovery of a mouse mutant harboring a mutation in an adaptin protein required for vesicular trafficking, Ap1b1 (adaptor-related protein complex 1, beta 1 subunit). This mutant has laterality defects that precisely phenocopies those observed in the Dnah5 mutants [3]. Given the known roles of AP1B1 in recycling cargo destined for the membrane, and in regulating posttranslational modifications of secreted proteins, this suggests the intriguing possibility of a possible role for Ap1b1 in regulating NVP release at the node.

Fig. 39.5
Scanning electron microscopy (EM) images of mouse node. Scanning EM showing monociliated cells in the embryonic node of a wild-type (a) and Dnah5 mutant (b) mouse embryo. Note the markedly higher abundance of large vesicles that may correspond to an accumulation of NVPs in the Dnah5 mutant node (From Tan et al. [23])
39.8 “Two-Cilia” Hypothesis
Another model proposed to account for the role of nodal flow in left-right patterning suggests motile and primary cilia are both involved in the specification of left-right patterning. This model proposes that primary cilia in the perinodal crown cells on the left side of the node undergo mechanosensory transduction to detect nodal flow, resulting in the elevation of Ca2+ to the left of the node. The notion that primary cilia may mediate flow sensing was first postulated with the finding of left-right patterning defects in polycystin-2 (Pkd2) mutant mice [34], an ion channel protein known to be associated with polycystic kidney disease. Pkd2 is localized in nodal cilia [35], and Pkd2 mutant mice failed to show Ca2+ elevation with nodal flow, while the iv Dnah11 mutant mice with immotile cilia displayed left-right randomized calcium elevation. Together these observations support a role for polycystin-2 in mediating Ca2+ signaling upstream of Nodal signaling.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


