Fig. 36.1
Transgenic mouse model displaying transposition of the great arteries (TGA). 3D reconstructions from magnetic resonance imaging (MRI) data sets [1] (a, b) and hematoxilin and eosin-stained (H&E) sections (c, d) of E15.5 fetuses. (a, c) By E15.5, the heart and its associated great vessels have developed into the mature configuration with the aorta (Ao) arising from the left ventricle (LV) and the arterial duct (AD) arising from the right ventricle (RV). (b, d) The heart of a transgenic fetus mutated for transcription factor AP-2alpha (Tcfap2a) presents with TGA, whereby the aorta arises from the RV and the DA arises from LV. Note how the two arteries lie in parallel (b) rather than spiral around each other (a). The presence of a pulmonary artery (PA) joining the AD is indicated (d) as is a VSD (b). Other abbreviations: LCC left common carotid, LSA left subclavian artery, RCC right common carotid, RSA right subclavian artery. Scale, 500 μm
36.2 Left–Right Patterning
Despite many human syndromes presenting with some form of cardiovascular defect, TGA is extremely rare in any syndrome except for heterotaxy [2]. However, TGA may be sporadically associated with VACTERL syndrome. Transgenic mice with a deletion of a gene associated with this syndrome, proprotein convertase subtilisin/kexin type 5 (Pcsk5), have TGA and double-outlet right ventricle (DORV) [3]. Heterotaxy is a rare congenital defect where the major organs are positioned abnormally within the body (see Chaps. 37 and 38). In the normal body, the position of the organs is called situs solitus, but when the organs are found in the mirror image of normal, it is known as situs inversus. However, when the reversal of organs is incomplete, this is known as situs ambiguous and can have profound effects on the formation of the heart and lungs. Indeed, it is considered that defects in the establishment of the left–right axis almost always result in complex congenital heart malformations [4].
The establishment of laterality in the embryo is conducted through the asymmetric and transient expression of transforming growth factor (TGF) β-related proteins that are both positively and negatively regulated (Fig. 36.2). This pathway begins in the node, a pit at the end of the primitive streak containing posterior-tilting ciliated cells that rotate. In the early mouse embryo (around E7.5 of gestation), these rotating cilia cause a leftward nodal flow that initiates the left–right axis determination process. In a mutant mouse line known as iv/iv (inversus viscerum), half of all these mice present with situs inversus, associated with complex cardiac malformations, including TGA [6]. In iv/iv embryos, the nodal cilia are immotile because of a mutation in the gene encoding the motor protein, axonemal dynein heavy chain 11 (Dnah11) [7]. The nodal flow is therefore absent resulting in randomized laterality of visceral organs [8, 9].
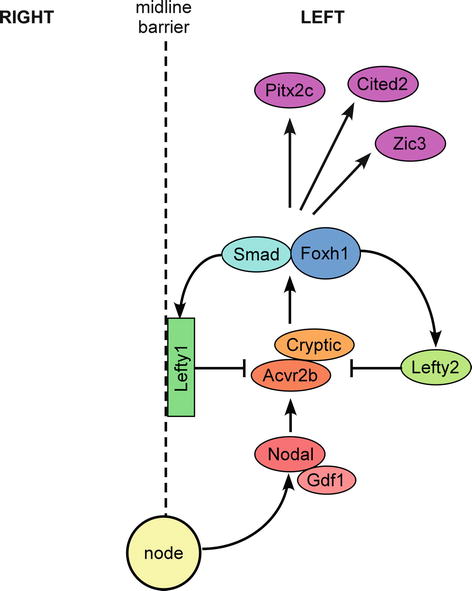
Fig. 36.2
Signaling molecules involved in the establishment of laterality. Nodal is expressed in the node from where it is transferred to the left lateral plate mesoderm (LLPM) forming a growth differentiation factor 1 (Gdf1)–Nodal heterodimer. Nodal signals through type I and type II transforming growth factor (TGF) β receptors (e.g., activin A receptor, type IIB; Acvr2b) and the co-receptor Cryptic. Smad proteins, along with forkhead box H1 (Foxh1), transduce the Nodal signal, and in response, left–right determination factor (Lefty) 1 and Lefty2 are expressed asymmetrically in the LLPM. The Lefty proteins inhibit Nodal signaling by competitively interacting with Cryptic and the type II TGFβ receptor, thereby acting as antagonists of Nodal and restricting its expression to the LLPM. The Nodal signaling pathway results in the induction of genes involved in left–right patterning, e.g., paired-like homeobox 2c (Pitx2c), Cbp/p300-interacting transactivator, with Glu/Asp-rich carboxy-terminal domain, 2 (Cited2) and Zic family member 3 (Zic3) (Adapted from Shiratori et al. [5])
The cilia found in the node are localized on the posterior portion of the apical surface of each of the node’s cells and display a morphological planar polarization. This planar cell polarity (PCP) regulates the polarity of epithelial cells in relation to their apical–basal axis [10], and PCP proteins are responsible for regulating planar polarity within the node [11]. As the polarity and function of the nodal cilia are critical for establishing the left–right signaling pathway, it is perhaps not surprising that left–right patterning defects leading to TGA can be found in some transgenic mouse models mutated for PCP genes. For example, half of all mice deficient for the PCP gene Dishevelled 2 (Dvl2) die from defects in outflow tract development that include TGA, DORV, and common arterial trunk [12] (More information on common arterial trunk is provided in Chap. 48). The defects observed in Dvl2 mutant mice are more penetrant in mice doubly homozygous for Dishevelled 1 and Dvl2. Cardiac outflow tract defects are also seen in mice mutated for Scrib (Circletail, Crc), another PCP gene [13]. The defects observed include TGA, as well as DORV, aberrant right subclavian artery, and atrioventricular septal defect.
Nodal is expressed in the node where it is subsequently transferred to the left lateral plate mesoderm (LLPM). In transgenic mice specifically lacking Nodal activity within the node, left–right patterning defects occur affecting the location of the stomach, right pulmonary isomerism, and heart looping randomization resulting in TGA [14]. Growth differentiation factor 1 (Gdf1) is co-expressed with Nodal in the node and is essential for Nodal expression in the LLPM, forming a Gdf1–Nodal heterodimer. Gdf1 is closely related to the Xenopus Vg1 gene, which influences the left–right patterning of the heart and viscera in the frog [15]. Although mouse Gdf1 and frog Vg1 proteins have limited sequence homology, the two proteins have functional and regulatory similarities [16]. In mice, Gdf1 is expressed in the primitive node, the ventral neural tube, and the LLPM of developing mouse embryos. Loss of Gdf1 is associated with the incorrect formation of the left–right axis, affecting the location of the stomach, spleen, pancreas, intestines, kidneys, and adrenal gland, and right pulmonary isomerism is also observed. Gdf1-null embryos are not viable, with only two-thirds of mutants surviving until birth, which then die within the immediate perinatal period. Mutant embryos present with cardiovascular defects including TGA as well as ASD, VSD, or a common atrioventricular canal [17]. Loss of Gdf1 results in the absence of left–right determination factor (Lefty) 1 in the left side of the floor plate and Lefty2 and Nodal in the LLPM suggesting that Gdf1 is likely to be directly or indirectly upstream of these genes [17]. A zebra fish model has been used to demonstrate the functional consequences of GDF1 mutations found in human patients with congenital heart disease, including TGA, tetralogy of Fallot, DORV, and interrupted aortic arch [18] (see Chap. 42 for more information on interruption of the aortic arch). These experiments showed that each of the mutations were either hypomorphic or resulted in a complete loss of function of Gdf1.
Expression of Gdf1 in perinodal cells requires sonic hedgehog (Shh) signaling, since Gdf1 expression is lost in the Smoothened (Smo) mutant mouse [19]. Smo is an essential component of the hedgehog signaling pathway. Downregulation of Gdf1 expression, which is also observed in transgenic mice with aberrant ciliogenesis [20], likely accounts for the lack of Nodal expression in the LLPM in these mutants with impaired hedgehog signaling. Moreover, transgenic mouse experiments have shown that Shh signaling in the ventral foregut endoderm is required for cardiac morphogenesis at the arterial pole of the heart as mice with a conditional deletion of Smo from the second heart field also display TGA [21].
Nodal is a ligand that can signal through type I and type II TGFβ receptors, one of which is the activin receptor, Acvr2b. Mice lacking Acvr2b have left–right patterning defects and TGA [22]. Activins are dimeric growth and differentiation factors which belong to the TGFβ superfamily. Cryptic, an EGF–CFC (epidermal growth factor–Cripto-FRL1-Cryptic) family protein, is a Nodal co-receptor, and transgenic mice lacking Cryptic have left–right patterning defects and cardiovascular abnormalities including TGA [23, 24]. Smad proteins, along with Foxh1, transduce the Nodal signal, and transgenic mice lacking Smad2 die early in development from defects in gastrulation [25]. However, transgenic mice doubly heterozygous for Smad2 and Nodal (i.e., Smad2 +/− ; Nodal +/− ) display TGA associated with right pulmonary isomerism of the lung, in more than 50 % of cases [25].
In response to Nodal signaling, Lefty1 and Lefty2 are expressed asymmetrically in the LLPM. The Lefty proteins inhibit Nodal signaling by competitively interacting with Cryptic and the type II TGFβ receptor, thereby acting as antagonists of Nodal and restricting its expression to the LLPM. Lefty1 is expressed at the midline and is thought to act as a physical barrier preventing the movement of left-sided proteins across to the right LPM. Transgenic mice lacking Lefty1 have left–right patterning defects, such as left pulmonary isomerism and misalignment of the cardiac outflow tract resulting in TGA, as well as DORV [26]. Other cardiovascular malformations include VSD, ASD, and common atrioventricular canal. Mice null for Lefty2 die early during embryogenesis, but this can be overcome through the creation of an alternative allele in transgenic mice. Deletion of the Lefty2 asymmetric enhancer element prevents these early gastrulation defects, but also prevents the asymmetric expression of Lefty2 in the LLPM at the early somite stage [27]. In these mutant mice, visceral situs abnormalities were identified along with cardiovascular defects including TGA, as well as DORV and aberrant great arteries.
The Nodal signaling pathway in the LLPM results in the induction of genes that, when mutated in transgenic mice, result in left–right patterning defects. One of these genes important for establishing a left-sided identity within the developing embryo is Pitx2c. The paired-like homeobox transcription factor gene, Pitx2, has four known isoforms: Pitx2a and Pitx2b are expressed symmetrically in the brain, whereas the Pitx2c isoform is expressed asymmetrically in the LLPM [28, 29]. PITX2D has only been identified in humans [30]. Pitx2c plays a major role in the establishment of left–right asymmetry of the body and is a direct target of Nodal [31]. Transgenic mice null for Pitx2c display outflow tract defects with rotational anomalies, leading to TGA [32]. This suggests that embryonic laterality affects outflow tract rotation and consequently may be the cause of some instances of congenital heart disease such as TGA.
Other genes downstream of the left–right signaling pathway include Cbp/p300-interacting transactivator, with Glu/Asp-rich carboxy-terminal domain, 2 (Cited2) and Zic family member 3 (Zic3). The Cited2 null mouse presents with left–right patterning defects and cardiovascular abnormalities including TGA [33, 34], as do mice null for Zic3 [35]. Indeed, mutations in ZIC3 have been found in patients with heterotaxy [36].
TGA has also been identified in transgenic mouse mutants not necessarily directly linked to a left–right patterning defect. These include mice mutated for Perlecan, a heparan sulfate proteoglycan [37], in which a high incidence of TGA is seen in fetuses with an intact ventricular septum but in the absence of any other laterality defect. It appears that TGA in this model is a consequence of excess mesenchyme preventing proper outflow tract rotation and thereby producing discordant ventriculoarterial alignment. Other such examples are the removal of PR domain-containing protein 1 (Prdm1) from the second heart field [38] and transgenic mice null for the endothelin A receptor (Ednra), which display craniofacial, aortic arch and outflow tract defects, which include TGA [39]. Mice mutated for fibroblast growth factor 8 (Fgf8) [40, 41] also display TGA, and in the chicken, TGA can be induced by blocking Fgf8 signaling with an Fgf8 receptor antibody [42]. This prevents adequate myocardium being added to the outflow tract resulting in a malalignment of the arterial pole.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


