Adhesion molecule
Alternative designation
Localisation
Ligand
Function
Basal expression
Selectin family
P-selectin
CD62P
Endothelial cell
L-selectin
Rolling
Yes
PADGEM
Platelet
PSGL-1
GMP-140
120 kD PSL
E-selectin
CD62E
Endothelial cell
L-selectin
Rolling
Noa
ELAM-1
CLA, SSEA-1
250 kD ESL-1
Ig-supergene family
ICAM-1
CD54a
Endothelial cell
CD11a/CD18
Adherence/transmigration
Yes
Leukocyte (myocyte, haematopoietic cell, fibroblast) [13]
CD11b/CD18
VCAM-1
CD106
Endothelial cell (myocyte, fibroblast, follicular dendritic cell) [14]
CD49d/CD29
Adherence
No/very low
PECAM-1
CD31
Endothelial cell
PECAM-1
Adherence/transmigration
Yes
Leukocytes
Platelet
MAdCAM-1
Endothelial cell
L-selectin
Adherence/transmigration
Yes
(Intestine)
CD49d/β7
Stimuli for the up-regulated expression of endothelial adhesion molecules include tumour necrosis factor, interleukin-1β, γ-interferon, substance P and bacterial endotoxin lipopolysaccharide [18]. Maximal endothelial adhesion molecule expression is generally noted between 3 and 6 h following exposure to the stimulus [7].
Transcription factors such as NF-κB and AP-1 have been shown to regulate the expression of endothelial adhesion molecules. NF-κB also leads to further production of inflammatory mediators, such as tumour necrosis factor, by the recruited leukocytes. Thus a vicious cycle occurs at the site of inflammation with inflammatory mediator induced endothelial adhesion molecule expression, leading to leukocyte recruitment, leading to further production of inflammatory mediators from the recruited leukocytes, leading to further endothelial adhesion molecule expression and leukocyte recruitment [7, 19].
There are species [20] and tissue [16] differences in endothelial adhesion molecule expression. More recently, genetic polymorphisms of endothelial adhesion molecules have been associated with their altered expressions in inflammatory conditions, such as atherosclerosis, rheumatoid arthritis, other connective tissues diseases, vasculitis and diabetes mellitus [20].
The up-regulated expression of endothelial adhesion molecules may be used as molecular markers of endothelial activation or inflammatory activity in a number of important cardiovascular and non-cardiovascular diseases [1, 31–33]. Furthermore, imaging of a series of endothelial adhesion molecules may be relevant because the expression profile (combination and level) of individual adhesion molecules may differ in different types and/or stages of diseases e.g., acute vs chronic vs resolving inflammation. For example, there is evidence that E- and P-selectin are involved in the selective recruitment of T-helper 1 cells [22, 23]. In myocarditis, T-helper 1-dominated responses are associated with the induction of myocarditis, whilst T-helper 2-dominated responses are associated with enhanced recovery [21]. Thus it may be possible to determine the status of myocarditis without tissue biopsy, by non-invasively imaging the endothelial adhesion molecule expression profile using quantitative ultrasound molecular imaging.
24.1.1.1 E-Selectin
Classically, E-selectin is expressed only on activated endothelial cells in the post-capillary venules. Its basal expression in resting endothelial cells, where present, is very low or undetectable. However, detectable low levels of constitutive E-selectin expression has been reported in some vascular beds of certain animals in the absence of overt inflammation [15, 16]. In addition to the post-capillary venules (the classical site of E-selectin expression), E-selectin expression has also been found in inflamed myocardial microvasculature [24], renal glomeruli [10] and the aorta [25, 26], in mouse models of Coxsackievirus B3 myocarditis [24], glomerulonephritis [10], and atherosclerosis [25], respectively.
E-selectin interacts with N- and O-glycans that contain the sialyl Lewis x tetrasaccharide or its isomers, such as the glycoprotein L-selectin (in man, not mice), E-selectin ligand-1 (ESL-1) and P-selectin glycoprotein ligand-1 (PSGL-1). These E-selectin ligands are present on neutrophils, monocytes, lymphocytes, myeloid cells, dendritic cells, and eosinophils [15].
E-selectin on the endothelial cell surface may be cleaved, releasing soluble E-selectin that lacks the intact cytoplasmic domain. The normal blood level of soluble E-selectin has been reported to be in the range of 0.1–3 ng/ml [12]. Cell-surface E-selectin may also be internalised into the endothelial cell by endocytosis [27].
E-selectin has been detected in human tissues of cardiac transplant rejection [28], myocarditis [29], dilated cardiomyopathy [29], coronary atheromatous heart disease [30], rheumatoid arthritis [31], inflammatory bowel disease [8], chronic inflammatory disease of the skin [9], cytomegalovirus infection [11] and sepsis [12].
24.1.2 Microbubbles as Ultrasound Contrast Enhancement Agents
24.1.2.1 Ultrasound Imaging for Microbubble Detection
Microbubbles are strong scatterers of ultrasound, readily detectable at typical clinical ultrasound frequencies of 2–13 MHz, despite being <6–10 um in diameter which is significantly smaller than the resolution of ultrasound. The echogenicity of the bubbles is dependent on the bubble size (size distribution), compressibility of the bubbles or gas, shell thickness/elasticity/viscosity, bubble scattering: attenuation ratio, insonating ultrasound frequency and power.
Microbubbles are compressible and may undergo non-linear radial oscillation in an acoustic field generating strong backscattered signals that can be differentiated from tissue signals using special non-linear ultrasound imaging modes (e.g., pulse inversion, Contrast Pulse Sequencing, Power Modulation).
In the acoustic field, microbubbles have different responses depending on the insonating acoustic power (peak negative pressure or mechanical index (MI)). At very low acoustic powers (e.g., <0.1 MPa or MI <0.1), bubbles oscillate linearly (contraction and expansion are similar in amplitude). At low-moderate powers (e.g., 0.1–1 MPa or MI 0.1–1), bubble expansion is larger than contraction (non-linear oscillation), and produces signals whose frequencies are multiples (harmonics) of the incident ultrasound wave. At high powers (e.g., >1 MPa or MI >1), the bubbles are destroyed rapidly, producing a brief non-linear response and harmonic signal of high amplitudes.
Non-linear imaging methods allow sensitive and specific detection of bubbles by evaluating the frequency components due to nonlinear response of the bubbles and suppressing those due to linear response of the tissues. Methods include high or low acoustic power (MI) settings (see below). High acoustic power settings are usually >1 MPa (MI >1), while the low power settings are usually 0.1–0.25 MPa (MI 0.1–0.25). The MI used for contrast ultrasound studies in clinical practice ranges 0.05–1.9.
The high acoustic power imaging modes (0.8–2.5 MPa or MI >0.8) are based on harmonic B-mode, harmonic Doppler or Power Doppler techniques. They cause rapid bubble destruction, associated with a brief emission of intense non-linear bubble signals, giving high bubble sensitivity. These bubble destructive imaging modes exhibit a higher signal-to-noise ratio, better bubble-to-tissue contrast and lesser nonlinear signal propagation in tissues compared with fundamental B-mode or Doppler imaging. However only intermittent single-frame contrast ultrasound images can be acquired, due to the rapid bubble destruction. This precludes continuous real-time imaging/assessment.
The low acoustic power imaging modes (0.05–0.5 MPa or MI 0.05–0.5) are based on pulse inversion (harmonic or Doppler), multi-pulse or coded excitation techniques. They cause minimal bubble destruction, allowing continuous real-time imaging. However, the lower acoustic power used has higher attenuation and lower bubble backscatter signal intensity, which reduces bubble detection sensitivity compared with the high acoustic power techniques. In harmonic mode, the attenuation is higher for returning harmonics than for the incident pulse. This effect cannot be compensated for by a higher amplitude of the incident pulse because it would give rise to: (a) nonlinear signal propagation in tissues, reducing signal-to-noise ratio and bubble-to-tissue contrast; (b) increased bubble destruction. Newer imaging modes based on multi-pulse sequence or coded excitation have increased signal-to-noise ratio and bubble-to-tissue contrast, allowing highly sensitive and specific bubble imaging at low acoustic powers. E.g., Contrast Pulse Sequencing [74] and Power Modulation imaging [75].
In practice, the relationship between the backscatter signal intensity and bubble concentration is exponential, with an approximately linear-range at low bubble concentrations [1, 76, 77]. At higher bubble concentrations, the increase in signal intensity plateaus; and at even higher concentrations, the signal intensity decreases because of: (a) signal saturation due to the limited dynamic range of the ultrasound system; and (b) signal attenuation due to multiple scattering effect [78], absorption, other ultrasound energy dissipating mechanisms such as viscous loss and thermal damping [79].
24.1.2.2 Microbubble Stability
Bubbles decay by dissolution (gas deflation), resulting in a decrease in size and number, and contrast ultrasound signal intensity. Factors influencing the stability of bubbles include the bubble size, characteristics of the bubble gas (low solubility, high molecular weight, low Ostwald coefficient, high saturated vapour pressure or low lipophilicity gives more stable bubbles), diffusivity of the gas across the shell, the shell thickness, surface tension, and solubility (bubbles with water soluble galactose shell last for shorter periods than those with relatively insoluble albumin- or phospholipid-shell), shell stability against mechanical perturbation, and the presence/absence of mechanical perturbation such as ultrasound insonation, shear force and systemic pressures in vivo [1, 36–39].
24.1.2.3 Biodistribution and Elimination of Microbubbles In Vivo
Microbubbles are considered as flow tracers which remain within the intravascular space and have rheology similar to that of the red blood cells [35]. The lungs and reticuloendothelial system (RES) are their major routes of elimination. They differ from iodinated or other contrast agents commonly used in scintigraphy, CT, MRI or PET imaging, which diffuse into the interstitium or cells, with often the kidneys as one of the major routes of elimination.
Following iv administration, the bubbles first reach the right heart, then the pulmonary circulation in the lungs, then return to the left heart and onwards to the systemic circulation. They continuously decay (decreasing in size and number) in the blood stream by gaseous deflation, the rate of which is influenced by the surrounding milieu for gaseous diffusion, such as the diffusion gradient and changes in the ambient pressure (such as in the ventricle, lungs or acoustic field) [36–39]. Iv administered bubbles undergo first-pass elimination in the lungs by size filtration, whereby those larger than the capillaries (e.g., >6–10 μm diameter) are mechanically trapped in the pulmonary microcirculation. The gas is exhaled out of the lungs unmetabolised. There is no significant sequestration of the bubbles in the lungs [40], presumably because the large trapped bubbles deflate rapidly. This first pass lung extraction has been shown to occur within 20–60 s [41, 42] in man following iv injection, and may decrease the total bubble gas volume by 30–50 %, depending on the size distribution of the bubbles administered. The bubbles are continuously removed from the circulation by RES uptake – primarily in the liver and spleen – involving phagocytosis by Kupffer cells or splenic macrophages, respectively [40, 43–48]. This is similar to other macromolecular contrast agents such as liposomes [49], supramagnetic iron oxide particles [50] and quantum dots [51]. The bubble shells are metabolised through pathways dictated by its component composition. Bubble elimination approximated to first-order kinetics (reviewed in Yeh et al. 2010 [1]).
In the pig, a significant proportion of microbubbles are removed by the pulmonary intravascular macrophages lining the lung capillaries [46, 52]. Pulmonary intravascular macrophages are only found in certain species such as cats and some mammals of the order Artiodactyla, which includes pigs, sheep, goats, and cattle. They are absent or negligible in rodents, rabbits, chickens, monkeys or man [53, 54]. The uptake of bubbles by the pulmonary macrophages causes powerful pulmonary vasoconstriction – apparently mediated by thromboxane [55]. An important implication of this is that experiments using certain animals (e.g., pigs) would require pre-treatment with steroids or non steroidal anti-inflammatory drugs, in order to prevent adverse effects from pulmonary vasoconstriction induced by the bubbles. The experiments would need to be designed such that the critical region of the hypothesis being tested will not be affected by these variables.
The biodistribution or elimination of targeting microbubbles are seldom described, however they are expected to be similar to that of the non-targeting bubbles, with the exception that targeting bubbles accumulate additionally at site(s) of their targeted molecule expression [44, 45]. Unlike circulating bubbles, retained bubbles at target sites are eliminated primarily by passive and/or ultrasound mediated gaseous deflation [39]; RES-uptake or rapid gaseous deflation in the lungs do not occur unless the bubbles targeted molecules expressed in the RES or lungs, respectively. Retained bubbles in non-RES tissues may be phagocytosed and degraded by leukocytes [56], but this is not universal [1] and likely dependent on the bubble property (e.g., chemical composition, physico-chemical and biological properties), the biology of the targeted molecule, the targeted cell, tissue or disease process. The elimination of targeting bubbles approximates to first-order kinetics [1]. The half-life of retained bubbles is longer the greater its initial (maximum) concentration [1]. This may be due to a bubble-to-bubble protection phenomenon recently described in vitro [57] and in vivo [1]–as the distance between adjacent bubbles shortens at higher concentrations, there is: (i) increased acoustical interactions between adjacent bubbles causing mechanical responses such that the net diffusion of gas out of the bubble population is reduced; and/or (ii) reduced concentration gradient for gas diffusion out of the bubbles, due to increased gas saturation in the micro-environment surrounding them. Such phenomenon highlights an important pitfall of conventional acoustic quantification methods (see Sect. 24.2.1).
24.1.2.4 Composition and Preparation of Microbubbles
The chemical composition and processing condition of the microbubbles influence their physical chemistry and mechanical properties, thus affecting their stability, yield, charge, size distribution, acoustic response, degree of non-specific adherence/retention in tissues, elimination and toxicity in vivo. Some of these variables are inter-related [1, 58–63].
Non-targeting and targeting microbubbles, produced by the pharmaceutical industry and others, which demonstrated ultrasound imaging in vivo are shown in Table 24.2. Detailed information on the composition and processing conditions required for making them is generally lacking in the public domain. For phospholipid-based microbubbles, the phospholipids can be dispersed along with surfactant polyethylene glycol (PEG) stearate in an organic solvent. The solvent is evaporated and replaced with an aqueous buffer. Bubbles are then formed using shear mixing approaches [58], where the mixture is sonicated or agitated vigorously in the presence of the filling-gas. For making targeting bubbles, targeting ligands can be conjugated to bubbles after the bubble formation (Fig. 24.1); or they can be conjugated to one of the shell components (e.g., a phospholipid species) prior to bubble formation [60].
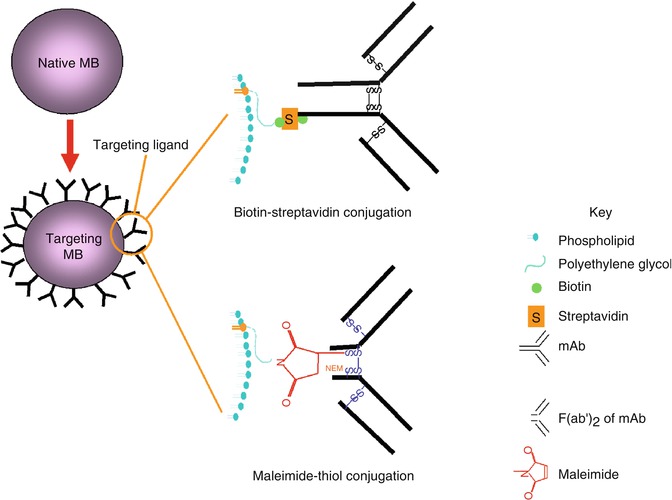
Table 24.2
Examples of microbubbles used in humans (albumin-, phospholipid- or other-shelled bubbles)
Microbubble | Composition | Gas | Net charge/zeta potential | Mean size or cumulative size distribution (μm) | Conc (x109/ml) | Application | Clinical trial status | Approval status | Developer |
|---|---|---|---|---|---|---|---|---|---|
BY963 | Phospholipid shell: contains DSPG | Air | LVO Doppler (cerebral, central and peripheral vessels) | Phase III (Europe) | Byk Gulden | ||||
Phase III | |||||||||
Definity (Perflutren, Aerosomes, MRX 115, DMP 115) | Phospholipid shell (8 nm thick): DPPA: DPPC: MPEG5000DPPE in 10:82:8 molar ratio | C3F8 | Neg | 1.1–3.3 98 % <10 μm | 1–1.5 | LVO | FDA-filed (USA and Europe), Phase III (Japan) | Licensed | Bristol-Myers Squibb (ImaRx Pharmaceuticals?) |
MP | Phase II/III | ||||||||
Hepatic and renal masses | FDA-filed (USA and Europe), Phase III (liver, Japan) | ||||||||
Breast masses | Phase II | ||||||||
Imagent (Imavist, AFO-150) | Phospholipid shell: contains DMPC | C6F14 | Neut | 5 | 0.5 | LVO | FDA-filed (USA) | Licensed | Alliance Pharmaceutical Corporation + Schering AG |
99.8 % <10 μm | MP | Phase II completed | |||||||
Stress echo (pharmacological stress) | Phase II | ||||||||
Lesions and blood flow abnormalities | Phase II | ||||||||
Prostate and breast | Phase II | ||||||||
Sonazoid (NC100100) | Phospholipid shell | C4F10 | Neg | 3 | 1 | LVO | FDA-submitted (USA) | Not approved | Nycomed Imaging |
MP | Phase II | ||||||||
Visualisation of liver lesions | Phase II completed | ||||||||
SonoVue (BR1) | Phospholipid shell: contains DSPC, DPPG, palmitic acid, PEG4000 | SF6 | Neg | 2.5 >90 % <8 μm | 0.1–0.5 | LVO | FDA-filed (USA and Europe) | Licensed | Bracco Diagnostics |
Macrovasculature | FDA-filed (USA and Europe) | Licensed | |||||||
MP | Phase II | ||||||||
BR55 (VEGFR2 targeting bubble) | Phospholipid shell: contains DSPE-PEG(2000), DSPE-PEG(2000)-peptide | C4F10 + N2 | 1.5 ± 0.1(SD), range 1–3 μm | 2 | Prostate cancer imaging | Phase 0 (results awaited) | Bracco Diagnostics | ||
Echovist (SH U454) | Galactose-based shell | Air | 99 % < 12 μm, 95 % < 8 μm | Licensed | Schering AG | ||||
Levovist (SHU 508A) | Galactose-based shell: 99.9 % galactose, 0.1 % palmitic acid | Air | Neg | 95 % < 10 μm | LVO | Marketed in Europe, etc. | Licensed (EU, Japan, Canada) | Schering AG | |
Doppler and CFI; vascular | Phase III completed in USA and Japan | ||||||||
Albunex (ABX) | Albumin shell (15 nm thick): | Air | −3 mV | 4 ± 2 (SD) | 0.5–0.8 | Withdrawn | Mallinckrodt | ||
5 % human serum albumin | |||||||||
Optison (FS069) | Albumin shell: albumin N-acetyltryptophan, caprylic acid | C3F8 | Slight neg | 2–4.5 | 0.5–0.8 | LVO | Marketed in USA and Europe | Withdrawn | Molecular Biosystems |
93 % <10 μm | MP | Phase II/III | |||||||
Stress echo | Phase II | ||||||||
Liver imaging | Phase II/III | ||||||||
TCD and kidney | |||||||||
Quantison | Albumin: | Air | 3.2 | 1.5 | MP | Phase II (suspended) | Not approved/withdrawn | Andaris Ltd | |
Human serum albumin (recombinant) | |||||||||
Cardiosphere (PB 127) | Bilayer polylactide/albumin shell | N2 | Slight neg | MP | Phase III completed | POINT Biomedical | |||
Sonovist (Sonavist, SHU 563A) | Polycyanoacrylate shell | Air | Liver, kidney, MP | Phase II | Schering AG | ||||
Acusphere (Imagify, AI 700) | Poly(lactic-co-glycolic acid) based shell | C4F10 | Neg | MP | Phase III | Acusphere Inc | |||
EchoGena (QW3600) | Charged surfactant (liquid droplet without shell): | DDFP | Neg | 3–10 | 3–5 | LVO | Approved for marketing in Europe | Withdrawn | Sonus Pharmaceuticals |
2 % DDFP emulsion containing surfactant (PEG Telomer B) + sucrose | MP | Phase II | |||||||
Stress echo (exercise and pharmacological stress) | Phase III completed | ||||||||
Chest pain in ER | Phase II | ||||||||
3D echocardiography | Phase III completed | ||||||||
Prostate |
Fig. 24.1
Strategy for converting native (non-targeting) microbubbles to targeting microbubbles for ultrasound molecular imaging. Native bubbles containing biotin or maleimide on its shell surface are conjugated with targeting ligands (e.g., mAb or its F(ab′)2 fragment). Top: traditional biotin-streptavidin conjugation where biotinylated mAb is linked to bubbles containing biotin on its surface, via streptavidin. Bottom: maleimide-thiol conjugation where reduced F(ab′)2 containing thiol (thiol produced from the reduction of interchain disulfide bond) is linked to bubbles containing maleimide on its surface. Abbreviations: mAb monoclonal antibody, NEM N-ethylmaleimide
Conjugation chemistries used in making targeting bubbles include non-covalent biotin-(strept)avidin and covalent maleimide-thiol [1, 64], pyridyldithio-thiol [102], carbodiimide [65], benzotriazole-amine [66], succinate-amine [103] or amine-succinimidyl [67] conjugations. To date, the biotin-(strept)avidin conjugation chemistry is the most commonly used, it being simple, robust, efficient, and forms very strong stable bonds. Although attractive for early developmental work, it is not suitable for human use because streptavidin or avidin is a bacterium Streptomyces avidinii protein or egg white protein, respectively, both known to produce unwanted immunogenic side effects [68]. An alternative maleimide-thiol conjugation chemistry has the advantage of low potential immunogenicity. Conjugate products containing maleimide have been tested in humans without significant immunogenic or toxic side-effects attributable to maleimide per se [69, 70]. This includes the maleimide-PEG-hemoglobin conjugate, Hemospan®, which has completed Phase III clinical trial [70]. The thioether bond formation between the maleimide on the bubble-shell and thiol on the targeting-ligand is strong and rapid at near neutral pH (bond strength in the order of nano newtons; second-order rate constant 0.8–1.2 × 104 M−1 s−1) [71]. The near neutral pH is advantageous in avoiding negative impact on the ligands and bubbles during the conjugation reaction, and in preventing dissociation of the ligands from the bubble-shell in vivo. In 2008, Yeh et al. reported E-selectin targeting bubbles, based on maleimide-thiol conjugation chemistry, that were effective for ultrasound molecular imaging in vivo (Figs. 24.2 and 24.3). These bubbles were stable, echogenic, specific to the targeted molecule and effective for real-time and quantitative ultrasound molecular imaging in vivo. Non-specific retention in non-RES tissues and adverse effects were minimal following iv administration [1, 34, 64]. Elsewhere, an amine-succinimidyl conjugation chemistry has been used in BR55 (a VEGFR2 targeting microbubble) [67], the only targeting bubble tested in humans so far – the results of its Phase 0 clinical trial are still awaited [72].
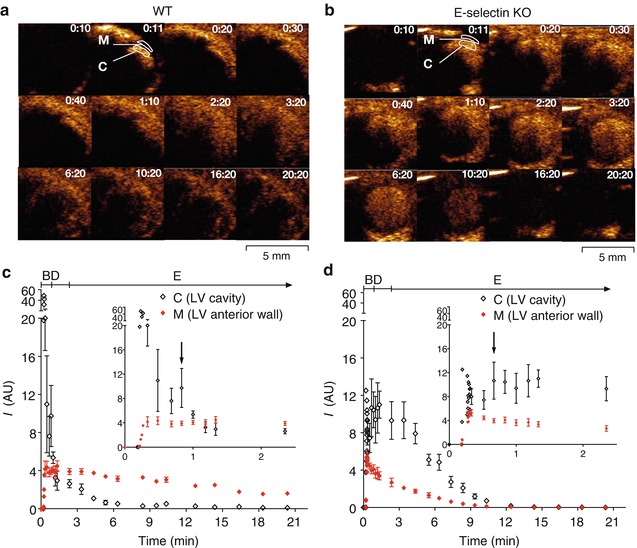
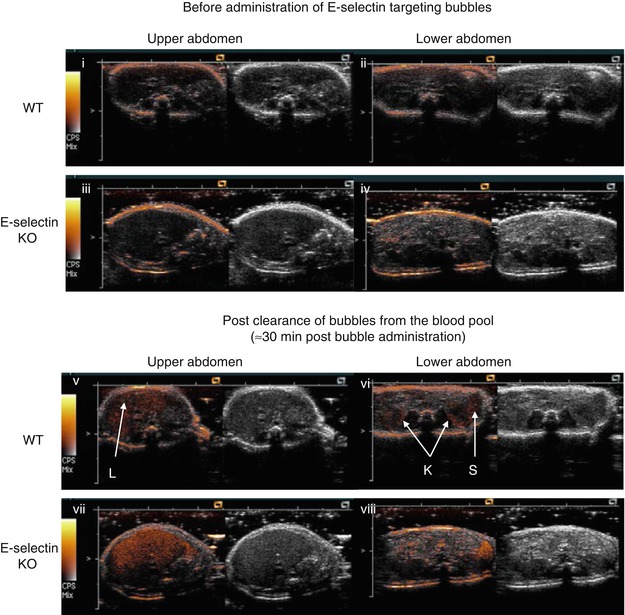
Fig. 24.2
Ultrasound molecular imaging of E-selectin expression in the mouse heart. Sequential Images (a, b). E-selectin targeting microbubbles were administered as a rapid iv bolus via the tail vein at 10s (0:10) to anaesthetised WT (a) and E-selectin KO (b) mice, both pre-treated with lipopolysaccharide to induce systemic inflammation involving the heart. Shown are sequential 14 MHz low MI Contrast Pulse Sequencing (a bubble-specific imaging mode) images of the heart in end-diastole, in parasternal short axis view of the LV. Bubble signal intensities are shown in a heated object scale (orange colour), tissue signals are not displayed. Results: Signals in the LV cavity and myocardium peaked within a few heart beats (<1 s). Signal attenuation in the LV cavity and myocardium decreased with time (e.g., frame 0:30 vs 6:20) as bubble concentrations decreased. Signal in the LV cavity (representing the concentration of free flowing bubbles in the blood pool) decreased more rapidly in the WT than the E-selectin KO mouse. Signal in the myocardium persisted after clearance of bubbles from the blood pool in the WT but not the KO mouse, indicating retention of the E-selectin targeting bubbles by E-selectin expressed in the WT myocardium. Time intensity curves of the LV cavity and myocardium in the WT (c) and E-selectin KO (d) mice. Plotted are the background subtracted mean acoustic units (AU) ± SD for the LV cavity (region of interest (ROI) C) and myocardium (ROI M). Suggested bolus (B), distribution (D) and elimination phases (E) of the bubbles are shown. A second lower peak signal intensity ≈ 40–50s post bubble administration could be observed in most animals (arrow, inset) as ultrasound signal attenuation reduced with decreased bubble concentration in the distribution phase
Fig. 24.3
Ultrasound molecular imaging of E-selectin expression in the mouse kidneys. WT and E-selectin KO mice were both pre-treated with lipopolysaccharide to induce systemic inflammation involving the kidneys. Baseline transverse images of the abdomen were obtained by scanning in a cranial-caudal direction, before iv bolus administration of E-selectin targeting microbubbles (i–iv). Imaging of the abdomen was resumed at ~30min post bubble administration (v–viii). Shown are 14 MHz low MI Contrast Pulse Sequence images of the upper abdomen encompassing the liver (i, iii, v, vii) and lower abdomen encompassing the kidneys (K) and spleen (S) (ii, iv, vi, viii). Bubble signal intensities are displayed in a heated object scale (orange colour) superimposed with tissue signals in grey scale. Results: The elimination of the circulating bubbles by RES-uptake could be inferred from the persistence of bubble signals in the liver and spleen, in both the WT and E-selectin KO mice, following the clearance of circulating bubbles from the blood pool. Specific targeting of the bubbles to E-selectin in the kidneys was observed in the WT, shown as the persistence of bubble signals in the kidneys after clearance of the circulating bubbles from the blood pool, which occurred in the WT but not E-selectin KO mice. The targeted bubble signal for E-selectin was located predominantly in the renal cortex, as expected (E-selectin was expressed predominantly on the endothelial cells of the renal glomeruli, which are concentrated in the renal cortex)
The number of targeting ligands per bubble ranged 0.3–80 × 104 (0.6–300 × 103 ligands/μm2), most in the order of 1–10 × 104/bubble (1–10 x 103 ligands/μm2) Table 24.3.
Table 24.3
Phospholipid-shelled targeting microbubbles tested for ultrasound molecular imaging in vivo
Origin (bubble ID, where available) | Conjugation chemistry | Bubble shell composition | Bubble gas | Targeting ligand | Targeting ligand density on bubble (conjugation reaction ratio) | Bubble diameter (μm) | Targeted molecules | Disease model | Tissues imaged | Host & bubble dose (number of bubbles) | Applicationa | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
UVA | Biotin- streptavidin | DSPC (83 mol %) PEG40-stearate (16 mol %) DSPE-PEG2000- biotin (1 mol %) ±DiI, DiO or rhodamine DHPE (small unspecified amount) | C4F10 | mAb Peptide Protein | 2–165 k/μm2 60–300 k/bubble | Mean: 1.9–3.4 Range: 94–97 % are <5 | α5-integrins αv-integrins β1-and β3-integrins CX3CR-1 H-2Kk (mouse MHC class I H-2Kk protein) on transfected bone-marrow derived endothelial progenitor cell ICAM-1 VCAM-1 MAdCAM-1 P-selectin Platelet glycoprotein-1bα Selectins (not discriminating amongst E-, P-, and L-selectin) vWF (A1-domain) | Angiogenesis (chronic ischemia, stem cell therapy, tumor) Coronary thrombosis & angioplasty Inflammation (atherosclerosis, cardiac transplant rejection, Crohn’s disease (small bowel), ischemia- reperfusion injury) | Aorta Bowel Heart Kidney Skeletal muscle Tumor (implanted in brain or flanks) Matrigel plug (implanted in abdomen) | Mouse: 1–10 × 106 Rat: 2.5–100 × 106 Dog: 108 | RT SQ | Lindner JR et al., Circulation 2001;104:2107 Ellegala DB et al., Circulation 2003;108:336 Leong-Poi H et al., Circulation 2003;107:455; Circulation 2005;111:3248 Weller GE et al., Circulation 2003;108:218; Cancer Res 2005;65:533 Sakuma T et al., Cardiovasc Res 2005;66:552 Bachmann C et al., Gastroenterology 2006;130:8 Villanueva FS et al., Circulation 2007;115:345 Kaufmann BA et al., Circulation 2007;116:276; Eur Heart J 2007;28:2011; J Am Soc Echocardiogr 2010;23:79; Arterioscler Thromb Vasc Biol 2010;30:54 Behm CZ et al., Circulation 2008;117:2902 Kuliszewski MA et al., Cardiovasc Res 2009;83:653 McCarty OJ et al., JACC Cardiovasc Imaging 2010;3:947 Carr CL et al., Arterioscler Thromb Vasc Biol 2011;31:2526 Davidson BP et al., J Am Coll Cardiol 2012;60:1690 Khanicheh E et al., Arterioscler Thromb Vasc Biol 2013;33:2187 Liu Y et al., Circ Cardiovasc Imaging 2013;6:74 Ryu JC et al., Circulation 2013;127:710 |
UVA | Biotin- streptavidin | DSPC (83 mol %) PEG40-stearate (16 mol %) DSPE-PEG3400- biotin (1 mol %) | C4F10 | mAb Protein Peptide | NA | Mean: 3.2 | Selectins (not discriminating between E- and P-selectin) VCAM-1 VEGFR2 | Angiogenesis (inflammation, tumor) Atherosclerosis | Aorta Tumor (implanted in hind limb) | Mouse: 1–10 × 106 | Not RT | van Wamel A et al., Proc IEEE Ultrason Symp 2007;961 (abstract) Khanicheh E et al., PLos One 2013;8:e58761 |
UVA (bubbles loaded with plasmid DNA) | Biotin- streptavidin | DSPC (70 mol %) PEG40-stearate (13 mol %) DSPE-PEG2000- biotin (1 mol %) DSTAP (16 mol %) cDNA (plasmid containing firefly luciferase gene): 0.04 pg loaded per bubble ±DiI or DiO | C4F10 | mAb | NA | Mean: 2.5–3.1 | P-selectin | Microbubble targeted gene delivery in ischemia (ischemia- reperfusion) | Skeletal muscle | Mouse: 2 × 108 | Not RT | Xie A et al., J Am Coll Cardiol Img 2012;5:1253 |
Targeson (TargeStar-series) | Biotin- streptavidin | TargeStar/TargeStar-B Biotin coated bubbles Component details not disclosed TargeStar-SA Streptavidin coated bubbles, containing 2.7 k streptavidin/μm2 (streptavidin on distal tip of PEG) Component details not disclosed | Perfluorocarbon (not specified) | mAb Peptide scFvs | (Reaction ratio: ligands at >20x excess necessary to saturate streptavidin binding sites) | Mean: 1.9–2.5 Range: 1–8; >98 % are <8 | αvβ3-integrin αIIbβ3 (GPIIb/IIIa) CD147b EGFRb P-selectin VEGFR2 | Angiogenesis (tumor) Thrombosis Tumorigenesis | Artery (carotid) Tumor (prostate) Tumor (implanted in flank, hind limb or mammary fat pad) | Mouse: 0.84–100 × 106 | RT SQ | Rychak JJ et al., Mol Imaging 2007;6:289 Xuan JW et al., Mol Imaging 2009;8:209 Hu X et al., Invest Radiol 2012;47:398 Sorace AG et al., J Ultrasound Med 2012;31:1543 Knowles JA et al., Arch Otolaryngol Head Neck Surg 2012;138:662 Wang X et al., Circulation 2012;125:3117 |
Targeson (TS-02-008, bubbles loaded with plasmid DNA) < div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|