Figure 2.1
Histology showing remodelled pulmonary arteries in idiopathic pulmonary arterial hypertension (iPAH). Photographs of paraffin-embedded haematoxylin and eosin-stained human lung sections from patients with iPAH following lung transplantation. Medium sized pulmonary arteries with medial hypertrophy and intimal proliferation (a) and plexiform lesions (b) (Images kindly supplied by Dr Peter Dorfmuller, Paris)
Pathogenesis of PAH
It is likely that the pathogenesis of PAH is a multi-hit phenomenon, similar to that described in cancer biology. As such there is evidence for an underlying genetic predisposition and known “hits” such as increased blood flow (Eisenmenger Syndrome), auto-antibodies (connective tissue disease), exposure to drugs (such as appetite suppressants), viruses (HIV) and inflammation. PAH is considered to be a progressive disease, although the exact rate of progression and the exact order of events remain unknown. Most authorities consider that endothelial damage and resulting endothelial dysfunction is the initial hit. This is characterised by an imbalance of vasoactive hormone production and loss of endothelial barrier integrity, leading to exposure of circulating mediators to the underlying smooth muscle. Activated smooth muscle cells may then respond by proliferation and autocrine production of mediators such as ET-1, which may propagate the process.
Endothelial Dysfunction
The following section will describe the abnormalities seen in the vasoactive hormones ET-1, PGI2 and NO associated with endothelial dysfunction.
ET-1
ET-1 is both a vasoconstrictor and mitogen that is mainly produced by the endothelium. However, under inflammatory conditions, the vascular smooth muscle is another important source. In patients with PAH there is increased production of ET-1 by the endothelium, and to a lesser extent smooth muscle, in remodelled vessels [6]. Furthermore, circulating plasma levels of ET-1 are raised in patients with PAH [7]and, importantly, increased circulating levels of ET-1 correlate with increased right atrial pressure, increased pulmonary vascular resistance, decreased pulmonary artery oxygen saturation and increased mortality [8–11].
NO
NO is a gaseous, lipophilic, free radical produced by the endothelium, which acts, via activation of soluble guanylate cyclase, to increase intra-cellular cyclic GMP. This, in turn, results in vascular smooth muscle relaxation (vasodilation) by altering intracellular Ca2+ concentrations. With relevance to vascular remodelling, NO also inhibits leukocyte adhesion, platelet aggregation, thrombus formation, and vascular proliferation [12, 13]. Reduced levels of endothelial nitric oxide synthetase (eNOS) are observed in the lungs of patients with idiopathic PAH [14]. There are several other molecular mechanisms explaining the decreased NO bioavailability in PAH including increased levels of endogenous competitive inhibitor of constitutive eNOS (such as asymmetric dimethylarginine (ADMA)), eNOS “uncoupling”, decreased l-Arginine levels and increased NO scavenging by haemoglobin and reactive oxygen species (ROS) [15, 16].
PGI2
The major active metabolite of arachidonic acid (AA), PGI2, is a critical endogenous regulator of vascular homeostasis. PGI2 is produced in vascular endothelial cells and acts on neighbouring vascular smooth muscle, as well as circulating platelets [17]. PGI2 is an agonist of adenylate cyclase and therefore a potent vasodilator with antithrombotic properties, acting via increase in cyclic AMP [17, 18]. An imbalance in AA metabolism has been demonstrated in patients with idiopathic PAH, with evidence for decreased PGI2 and increased thromboxane (TXA2) urinary metabolites [19]. Furthermore, expression of the key enzyme for PGI2, synthesis PGI2 synthase (PGIS), is reduced in pulmonary arteries of patients with PAH [20].
In the following sections we will describe the pre-disposing factors and secondary “hits” that may contribute to vascular remodelling.
Genetic Predisposition
Although idiopathic and heritable PAH are rare, an improved understanding of the genetics of heritable PAH (constituting up to 10 % of “idiopathic” PAH cases) has revealed pathobiological mechanisms that may be relevant to PAH associated with other, more common, conditions. Specifically, the genetic defect underlying the majority (80 %) of cases of heritable PAH was identified as heterozygous germ-line mutations in the gene encoding the bone morphogenetic protein receptor (BMPR)-II [21, 22], a member of transforming growth factor-beta (TGF-β) superfamily (Fig. 2.2). Similar mutations have been found in 20 % of sporadic cases of idiopathic PAH [23]. Segregation analysis of affected families demonstrates that the disease is autosomal dominant often with markedly reduced penetrance (as low as 20 %) [24]. Furthermore, it appears that the predominant genetic mechanism is haplo-insufficiency [25]. A reduction in BMPR II expression has now been observed in other, more common, forms of human PAH [26] and in animal models of PAH [27, 28]. Indeed, selective deletion of BMPR II in animal models results in PAH, although usually at levels milder than in human disease [29]. More recently mutations have been found in genes encoding other members of the TGF-beta superfamily, such as endoglin, ALK-1 and SMAD 8 [30–32]. Interestingly, in a few affected families, abnormalites have also been found in non-TGF-beta superfamily molecules such as caveolin-1 [33] and KCNK3 [34].
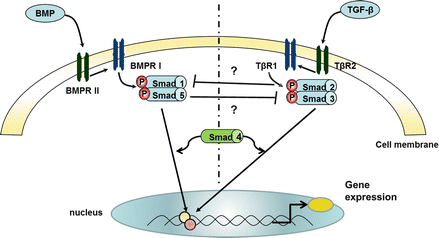
Figure 2.2
Schematic representation of bone morphogenetic protein (BMP) signalling via Smads and the interaction with the transforming growth factor (TGF)-β pathway. Abbreviations: BMP bone morphogenetic protein, BMPR BMP receptor, TGF transforming growth factor, TβR– TGF receptor; Smad– Mothers Against Decapentaplegic Homolog protein. Bone morphogenetic protein receptors (BMPRs) are members of the transforming growth factor-β (TGF-β) superfamily. Ligand binding to the BMPR I-II complex activates the signalling molecules Smad 1 and 5, which together with the co-Smad 4 regulate gene expression. In particular, it appears that in the healthy cell, most of the action of these Smads is to down-regulate cell proliferation genes and increase pro-apoptotic genes. BMPR II mutations, seen in patients with idiopathic PAH result in disrupted Smad 1, 5 signalling, and a relative increase in pro-proliferative/anti-apoptotic genes. It remains unclear how the BMPR I-II pathway interacts with traditional TGF-β signalling via Smad 2, 3
Blood Flow
Clearly increased flow is a key initiating event (and likely propagating factor) in the pathogenesis of Eisenmenger syndrome (ES), where increased left-to-right blood flow through a congenital shunt leads to perseverance of the fetal type (high pressure) pulmonary circulation. It is also known that turbulent, non-linear, flow leads to increased shear stress and activation of endothelial cells, through NF-κB dependent mechanisms [35–37]. Areas of turbulent flow are associated with formation of atherosclerosis, overlying thrombosis and vessel wall rupture in patients with ES [38].
There are several lines of evidence to suggest that BMPR II dependent signalling may be important in regulating responses to flow. Firstly, one of the ligands for BMPR II, BMP4 is involved in regulating inflammation and subsequent atherosclerosis production in endothelial cells [39, 40]. Endothelial cells release BMP4 in response to oscillatory flow, which in turn enhances monocyte adhesion. Co-release of endogenous inhibitors of BMP4, such as noggin and follicostatin, acts to attenuate this inflammatory pathway [41]. Secondly, BMPR II provides a site for multiple proteins to cluster in the plasma membrane, including cytoskeleton proteins, protein kinases (e.g., LIM kinase, Src, PKC, MAPKs) transcriptional factors (e.g., CtBP, NF κBp50) and transient receptor potential canonical 1 (TRPC1), a non-selective Ca2+ channel, some of which have been described as taking part in flow sensing mechanisms[42–47]. Interestingly, mRNA and protein/activity of TRPs are upregulated in lung and smooth muscle cells isolated from patients with idiopathic PAH (although the BMPR II status was unknown [48]). Thirdly, in the pig model of flow associated systemic-pulmonary shunt induced PH, concomitant to the rise of pulmonary vascular resistance and arteriolar medial thickness, there is a decrease in expression of BMPR II (and its co-receptor BMPR Ia). Conversely, pre-treatment with the angiotensin receptor antagonist, losartan decreased shunt-induced pulmonary vascular resistance and medial thickness with a return to normal of the BMPR II expression [49]. These data would suggest a close correlation of BMPR II function and shear stress response in vascular cells. However, this also raises an important question as to whether patients with BMPR II mutation are genetically more susceptible to aberrations in shear stress patterns generated by disturbed blood flow; BMPR II mutations could result in abnormality of intracellular response to blood flow, e.g., Ca2+ influx or intracellular Ca2+ release (TRPC1), cytoskeleton rearrangement (LIM kinase), and gene expression (PKC, Src, NF κB, MAPKs) or BMPs.
Drugs and Toxins
PAH is associated with a variety of drugs and toxins [50]. The most common class of drugs implicated is appetite suppressants, which include drugs such as dexfenfluramine. As these drugs are involved in serotonin signalling (see later section), it is likely that serotonin plays an important role in the pathogenesis of PAH associated with these drugs. However, there is an ever increasing list of drugs associated with the development of PAH (see Table 2.1), the latest being interferons [51]. The most well documented toxic insult (“toxic inflammatory PH”) occurred after ingestion of adulterated food oil. This led to acute lung injury associated with eosinophilia and myalgia [52]. PAH occurred in 20 % of hospitalized patients, 2–4 months from onset and in 8 % of longer-term survivors.
Table 2.1
Classification for drugs and toxins as risk factors for the development of PAH
Classification of risk associated with drug/toxin | Drug/Toxin |
|---|---|
Definite group | Aminorex Fenfluramine Dexfenfluramine Toxic rapeseed oil Benfluorex SSRIs |
Likely group | Amphetamines l-Tryptophan Methamphetamines Dasatinib |
Possible group | Cocaine Phenylpropanolamine St. John’s wort Chemotherapeutic agents Interferon α and β Amphetamine-like drugs |
Unlikely group | Oral contraceptives Estrogen Cigarette smoking |
Viruses and Parasites
Viruses
The prevalence of PAH in patients infected with HIV is estimated at around 0.5 % [53]. The onset of PAH in these patients confers a worse prognosis [54]. The precise mechanism by which HIV leads to pulmonary vascular remodelling is unknown, but it is likely to be a multifactorial process, and at least in part related to the induction of proinflammatory cytokines and growth factors from the induced chronic state of immune activation. Unsurprisingly, altered BMPR II signaling is likely to be involved [55], although germ-line BMPR II mutations are not usual in these cases [54]. HIV virus has never been detected in vascular lesions [56]; therefore, indirect action by HIV proteins is likely. For example, the envelope protein glycoprotein-120 (responsible for HIV binding and entry into macrophages) has been shown to induce apoptosis and increase ET-1 secretion from endothelial cells in vitro [57]. Furthermore, HIV-1 negative factor (nef) antigen, crucial for maintenance of the HIV viral load, has been localized to cells within complex vascular lesions in these patients [58], with a proposed mechanism being an increase in endothelial cells apoptosis followed by the emergence of apoptosis-resistant endothelial cells with a hyperproliferative phenotype [59]. Other viruses have also been implicated in the pathogenesis of PAH. For instance, genes coding for human γ herpes virus 8 (or Kaposi sarcoma-associated herpes virus) proteins have been detected in plexiform lesions [60]. In support of this observation, human γ herpes virus 8 infection of pulmonary microvascular endothelial cells in vitro results in an apoptosis-resistant cell phenotype [61], as well as a reduction in BMPR II expression [62]. Furthermore, chronic active Epstein-Barr virus infection has been associated with high circulating IL-6 levels in human, and with the development of PAH [63]. However, there is some controversy in these associations and it remains possible that there are geographical differences in possible infectious insults [64–66].
Parasites
Schistosomiasis is likely to be the most common cause of PAH worldwide: 200 million people are infected, and the associated prevalence is 2–5 % [67]. The development of PAH is thought to follow hepatosplenic infection with Schistosoma mansoni and the subsequent development of portal hypertension: after entering the skin, the fresh-water parasite migrates to the lungs and then to the portal venous system, where it matures [68]. The deposition of eggs in liver veins leads to presinusoidal granulomatous inflammation, peri-portal fibrosis, and portal hypertension. The resultant opening of portocaval shunts both increases pulmonary blood flow and creates a pathway for eggs to lodge in the pulmonary capillaries [67, 68]. Histologically, pulmonary vascular lesions are similar to those seen in idiopathic PAH, including the presence of plexiform lesions [69]. The development of schistosomiasis-associated PAH is thought to be due to the deposition of eggs in lung tissue causing mechanical vessel impaction and focal arteritis, inflammation relating to the formation of granulomas around the eggs, and increased pulmonary blood flow. The contribution of inflammation to vascular remodelling in this setting is not well understood. However, using murine models, it appears that a switch from a Th1 to a Th2 immune response is important [70, 71]. Although these models are important in enhancing our understanding of this globally important cause of PAH, the phenotype does not accurately reflect human disease in that there is less portopulmonary pulmonary hypertension.
Inflammation
Inflammation is present in all forms of PAH. However, how inflammation may contribute to the pathogenesis of PAH is not clear (Fig. 2.3.). It is possible that inflammation may initiate vascular remodelling (“initial hit”), be integral in its propagation (“secondary hit”), or just be a reactive response to ongoing remodelling (“bystander” phenomenon). Initial hits may include infections, drugs, or toxins (see previous sections). There may be a relationship between such an inflammatory hit and other factors such as BMPR II status [72], which may alter the subsequent inflammatory response. Whatever the exact relationship, there is evidence for activation of both the innate immune system (activation of macrophages/monocytes) and adaptive immunity (through specific T-cell and B-cell receptors). The cytokines and chemokines subsequently produced may propagate further inflammatory processes and, either on their own, or together with production of growth factors, drive vascular remodelling processes.
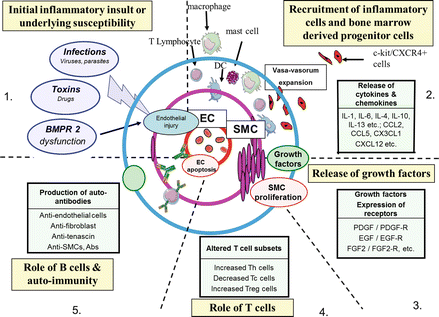
Figure 2.3
The role of inflammation on the pathogenesis of pulmonary arterial hypertension. It is likely that there are 5 stages to the influence of inflammation on pulmonary vascular remodelling: 1 Initial inflammatory insult and/or underlying genetic susceptibility; 2 Recruitment of inflammatory cells and bone marrow derived progenitor cells; 3 Release of inflammatory mediators by either vascular cells themselves or associated inflammatory cells; 4 Role of T cells-it is likely that T cells in some way regulate the inflammatory process; 5 Role of B cells and auto-immunity-there is increasing evidence for anti-bodies against key vascular elements driving the inflammatory process
Evidence for Inflammatory Cells
T Cells
T cells are essential components of the adaptive immune response. Several lines of evidence support a role for T cells in the development of PAH. In animal models, the athymic nude rat (without T cells) develops PAH more readily than those with intact T-cell production [73]. In the monocrotaline animal model, depletion of Th cells, as well as those of Th2, ameliorates the extent of PAH, again suggesting the importance of a Th2 antigen-driven immune response [74]. Whereas T-cell infiltrates are increased in patients with idiopathic PAH [75, 76], it appears that there is a decrease in CD8+ cytotoxic (Tc) cells and an increase in regulatory (Treg) cells [77]. The precise role of Treg cells in PAH is currently being investigated.
B Cells
B cells generate antibodies to specific antigenic epitopes. Levels of antinuclear antibodies are increased in patients with PAH [78], and autoantibodies directed against endothelial cells [79] and fibroblasts [80] have been described. These autoantibodies may play a role by inducing adhesion molecule expression [81] or inducing endothelial cell apoptosis [82], which may, in turn, contribute to the apoptosis-resistant phenotype. Finally, the development of PAH in patients with systemic sclerosis is seen in association with specific subsets of human leukocyte antigen alleles [83], although the significance of this remains uncertain.
Mast Cells
Accumulation of mast cells has been described in several types of PAH [84, 85]. A recent study has identified an increase in c-kit-positive cells (including mast cells) in remodelled vessels, as well as mobilization of bone marrow-derived circulating progenitor cells [86]. The increase in mast cell numbers consists mainly of the chymase-secreting subset, numbers of which correlate with the hemodynamic severity of the disease [85]. It is possible that secreted substances from mast cells may be directly vasoactive or have secondary effects such as increased production of matrix metalloproteinases, with consequent involvement in vascular remodelling [84, 87].
Monocytes/Macrophages and Dendritic Cells (DC)
Macrophages are the universal phagocytes that differentiate from tissue monocytes. Along with DC, they are professional antigen-presenting cells, displaying antigen bound to major histocompatibility complex class 2, ready for recognition by T cells. Increased numbers of all these cell types are present around remodelled vessels in PAH [88]. Furthermore, apart from being more numerous in patients with idiopathic PAH, these cells are more “activated” as determined by enhanced nuclear NF-kB immunohistochemical staining [89].
Endothelial Progenitor Cells
Endothelial progenitor cells (EPCs) were first discovered in adult human peripheral blood as a population of CD34 or kinase insert domain receptor (KDR)-positive peripheral blood mononuclear cells which were able to differentiate into mature endothelial cells and were involved in neo-vascularisation [90]. EPCs are likely to constitute a pool of circulating cells driven from bone marrow in response to stimuli such as tissue ischaemia and vascular damage, and contribute to vascular repair and postnatal neovascularization [91, 92].
Recent animal and clinic studies have suggested that EPCs may be involved in the pathogenesis and progression of PAH [92–94]. For instance, in the mouse hypoxia model of pulmonary hypertension, whole bone marrow transplantation of enhanced green fluorescent protein (GFP)-transgenic mice to lethally irradiated Chimeric mice resulted in the mobilisation of GFP+ cells to remodelling pulmonary arteries [95, 96]. Furthermore, in the lung from patients with PAH, an upregulation of progenitor cell markers such as CD133 and c-Kit in remodelled arteries is observed, especially in plexiform lesions [97]. In these patients, levels of circulating EPCs (CD34+CD133+VEGFR2+) were increased; however, the ability to form vascular networks from late-outgrowth EPCs (CD34+CD146+vWF+CD133+/−) was impaired [97].
Evidence for Cytokines/Chemokines
Cytokines
Interleukin (IL)-1-β
IL-1-β is a potent proinflammatory cytokine. In human PAH, serum concentrations are raised [98] and these correlate with a worse outcome [99]. IL-1-β is produced in larger amounts in the monocrotaline animal model, compared with the chronic hypoxic model of pulmonary hypertension [82]. Furthermore, repeated treatment with an IL-1 receptor antagonist reduces pulmonary hypertension and right ventricular hypertrophy in the monocrotaline model, although not in the chronic hypoxia counterpart [100].
IL-6
IL-6 is another proinflammatory cytokine synthesized by many cell types. Plasma concentrations of IL-6 are elevated in idiopathic PAH [98], and they correlate with severity of disease and with increased mortality [99]. In patients with systemic sclerosis, elevated circulating IL-6 concentrations predict the presence of associated PAH [101]. Elevated serum IL-6 concntrations also correlate with hemodynamic severity in pulmonary hypertension associated with COPD [102] and in other forms of PAH, including sickle cell disease-associated PAH [103]. Pulmonary IL-6 production is also increased in experimental pulmonary hypertension and is thought to reflect increased production by both inflammatory cells and vascular cells [104, 105]. In turn, IL-6 has many effects on inflammatory and vascular cells that may promote vascular remodelling. These include accumulation of perivascular T lymphocytes [106], stimulation of endothelial cells to produce chemokines [107], promotion of pulmonary artery smooth muscle cell and endothelial cell proliferation [99, 105].
IL-13
IL-13 is a cytokine secreted by many cells, especially Th2 cells and mast cells. It is important in forming granulomata in response to parasites (including schistosomiasis) and its effects on immune cells are similar to those of IL-4. Loss of IL-13 signaling reduces pulmonary vascular remodeling in models of pulmonary hypertension and its effects on T cells suggest an indirect role in regulating Th2 responsiveness [70, 74]. A relative increase in the negative-regulating decoy receptor, IL-13Rα2, is observed in pulmonary arterial smooth muscle cells from patients with idiopathic PAH compared with the active receptors (IL-13Rα1 and IL-4R), as well as in monocrotaline and hypoxic pulmonary hypertension models [108]. Perhaps surprisingly, IL-13 is antiproliferative to pulmonary arterial smooth muscle cells in vitro, with an associated reduction in ET-1 release [108]. Overall, these data suggest that dysregulated signalling of this Th2 cytokine is likely to contribute to vascular remodelling in PAH.
Chemokines
Monocyte Chemoattractant Protein(MCP-1)
MCP-1 (or CCL2) is a chemokine produced by vascular cells that stimulates monocytes/macrophage activation and migration, with actions mediated via the chemokine (C-C motif) receptor. Elevated levels of MCP-1 are found in the plasma and lung of patients with idiopathic PAH [109], although they do not correlate with disease severity. Furthermore, vascular smooth muscle cells and endothelial cells from patients with idiopathic PAH release high levels of MCP-1. In addition, vascular smooth muscle cells from patients with idiopathic PAH express increased levels of the chemokine (C-C motif) receptors, exhibit exaggerated migratory and proliferative responses to MCP-1, and these can be blocked by MCP-1 antibodies [109]. Interestingly, an anti-MCP-1 monoclonal antibody has been used in patients with rheumatoid arthritis in a randomized controlled trial, but not yet in PAH [110].
Regulated on Activation, Normal T-cell Expressed and Secreted (RANTES)
RANTES (or CCL5) is a chemokine that mediates the trafficking and homing of T lymphocytes, monocytes, basophils, eosinophils, and natural killer cells through different chemokine receptors [111]. Pulmonary RANTES mRNA is elevated in patients with PAH and shown to be of endothelial cell origin [5, 89, 111]. However, the role of RANTES in the pathogenesis and progression in PAH has not been elucidated.
Fractalkine (CX3CL1)
Fractalkine (CX3CL1) is a chemokine expressed as a soluble or as a membrane-bound form. The effects of fractalkine are mediated through the receptor, CX3CR1, expressed by many cell types. Elevated concentrations of soluble fractalkine are seen in patients with PAH [112], although the association of such elevation with disease severity and prognosis is not currently proven. Increase expression of fractalkine mRNA and protein were detected in pulmonary vascular endothelial cells. Furthermore, it has been shown that the CX3CR1 expression and function were upregulated on both CD4+ and CD8+ T lymphocytes in patient with PAH [89, 112]. It is likely that the concomitant increase of expression in ligands in endothelial cells and receptors on T cells may contribute to the perivascular inflammatory cell influx [113]. Fractalkine and CX3CR1 expression were also shown to be upregulated in pulmonary smooth muscle cells in the monocrotaline rat model of pulmonary hypertension. Finally, fractalkine treatment increased pulmonary arterial smooth muscle cell proliferation in vitro, indicating a direct role of fractalkine in vascular remodelling [113].
Platelet Derived Growth Factor (PDGF)
PDGF is secreted as a homodimer of genetically distinct but structurally similar polypeptides (chains A-D) [114]. Additionally, only chain A and chain B can form a functional heterodimer. Increased expression of PDGF-A, PDGF-B and PDGF receptor (R)-α, PDGFR-β mRNA and protein have been found in pulmonary arteries from transplanted PAH patients. Further study demonstrated that PDGF-A and PDGF-B proteins were localised in smooth muscle and endothelial cells, whilst PDGFR-α, PDGFR-β proteins predominated in smooth muscle cells in remodelled pulmonary arteries including plexiform lesions. In addition, intense staining of the ligands and receptors has also been observed in perivascular inflammatory infiltrates [115].
PDGF is a strong mitogen for cells of mesenchymal origin including smooth muscle cells and fibroblasts [114, 115]. Imatinib, a well-established tyrosine kinase inhibitor of the kinase BCR-ABL; the receptor for the stem cell factor, c-KIT; and the PDGFR [116], has been shown to have anti-proliferative and pro-apoptotic effects on pulmonary artery smooth muscle cells taken from patients with PAH in vitro [115], and has also been shown to reverse pulmonary vascular disease in animal models of PAH [117, 118]. See later section for clinical evidence.
Epidermal Growth Factor (EGF)
EGF promotes chemotaxis, mitogenesis and cytoprotection in epithelial and mesenchymal cells through binding to its receptor (EGFR). The EGFR belongs to the ErbB family of tyrosine kinase receptors [119]. EGF has been shown in vitro to increase pulmonary arterial smooth muscle cell proliferation [120]. In the monocrotaline animal model of pulmonary hypertension, the use of serine elastase inhibitors has been shown to reverse pulmonary vascular remodelling. In this study it was suggested that serine elastases, by breaking down extracellular matrix, could lead to the release of EGF and promote vascular remodelling [121].
Vascular Endothelial Growth Factor (VEGF)
VEGF is an endothelial cell specific mitogen and a potent angiogenic mediator. VEGF is produced by a variety of cells and has been implicated in physiological and pathological conditions associated with endothelial cell proliferation [122]. It has been suggested that VEGF is important in attenuating the development of PAH possibly by protecting endothelial cells from injury and apoptosis [123, 124]. Indeed, an overall decrease in pulmonary VEGF expression has been reported, in concert with a dramatic decrease in pulmonary vessel number and a significant increase in vessel wall thickness in the monocrotaline model of pulmonary hypertension [125]. Pulmonary VGEF and VEGF receptor were also down regulated in a rat model of pulmonary hypertension associated with idiopathic pulmonary fibrosis [124]. In contrast, serum VEGF levels are increased in systemic sclerosis patients with PAH [126]. Upregulation of VEGF has also been described in association with plexiform lesions, possibly representing an incomplete attempt at revascularization distal to an arteriolar occlusion [122, 127].
Fibroblast Growth Factor 2 (FGF2)
FGF2 is a member of a large family of heparin-binding growth factors. Increased lung and circulating FGF2 levels have been reported in both experimental and human pulmonary hypertension. Abnormally high levels of FGF2 were found in the blood of 51 % and in the urine of 21 % of patients with idiopathic PAH [128] and in 3 animal models: a lamb model of pulmonary hypertension developed by inserting an aortopulmonary vascular bypass graft [129] and the rat chronic hypoxic and monocrotaline models of pulmonary hypertension[130, 131]. It has been shown that excessive autocrine release of endothelial-derived FGF2 in idiopathic PAH contributes to the acquisition and maintenance of an abnormal endothelial cell phenotype in vitro and in vivo, enhancing proliferation through constitutive activation of ERK1/2 and decreasing apoptosis by increasing BCL2 and BCL-xL [132]. Finally, a recent study on apelin knockout mice showed that apelin deficiency led to increased expression of FGF2 and its receptor FGFR1 as a consequence of decreased expression of microRNA (miR)-424 and miR-503, which directly target FGF2 and FGFR1[133].
Serotonin (5-hydroxytryptamine, 5-HT)
Serotonin is an important vasoactive and mitogenic compound that is synthesised by endothelial cells and acts on pulmonary arterial smooth muscle cells [134]. The serotonin transporter (5-HTT) is required for serotonin to elicit its mitogenic effects and transgenic mice overexpressing 5-HTT have been shown to spontaneously develop PH. Serotonin receptors (5-HT2B) also appears to play important role in mediated vascular remodelling: mice with restricted expression of 5-HT2B receptors in bone marrow cells exacerbate hypoxia or monocrotaline-induced increases in pulmonary pressure and vascular remodeling, whereas restricted elimination of 5-HT2B receptors on bone marrow cells confers a complete resistance to the development of PH [135]. The role of serotonin system in the pathogenesis of PAH was further supported by the development of PAH in a sub-set of patients taking the anorexigenic drugs aminorex and dexfenfluramine. Both of these drugs are 5-HTT substrates and indirect serotinergic agonists [134]. The overexpression of 5-HTT in pulmonary vascular media may be caused by a polymorphism in the promoter region of the human 5-HTT gene which alters the level of transcription or, alternatively, by BMPR II dysfunction [136–139].
Evidence for Autoantibodies/Autoimmune Phenomenon
Anti-endothelial cell antibodies (AECAs) and anti-fibroblast antibodies (AFAs) have been detected in plasma from patients with idiopathic PAH and with systemic sclerosis-associated PAH (SSc-PAH) [79, 80]. AECAs and AFAs from patients with idiopathic PAH and SSc-PAH showed a distinct reactivity profile; AECAs from patients with idiopathic PAH bound more strongly to a 58 kDa band in dermal microvascular endothelial cells and to a 53 kDa band in lung microvascular endothelial cells. AECAs from patients with limited cutaneous SSc with or without PAH bound to two major bands (75 kDa and 85 kDa) in microvascular endothelial cells [79]. One the other hand, AFAs from these patients predominantly bound to 25-, 40-, and 60-kD protein bands [80]. Further study has identified 16 proteins: vimentin, calumenin, tropomyosin 1, heat shock proteins 27 and 70, glucose-6-phosphate-dehydrogenase, phosphatidylinositol 3-kinase, DAP kinase, and others as the target antigens of AFAs [140]. These proteins are involved in regulation of cytoskeletal function, cell contraction, oxidative stress, cell energy metabolism, and other key cellular pathways. Therefore autoantibodies may play an important role in PAH pathogenesis [140].
Current Treatment of PAH
As mentioned before the current treatment of PAH is based on endothelial dysfunction: hence endothelin receptor antagonists are used to counter increased ET-1 levels; phosphodiesterase type V inhibitors enhance endogenous NO levels; exogenous prostanoids replace deficiencies in PGI2 production. The evidence for and the use of these drugs is covered in depth in separate chapters. It is also worth noting that to improve safety and efficacy, a number of new drugs targeting these pathways have been tested in clinic trials and will be mentioned here.
Macitentan is a novel dual ET-1 receptor antagonist (ERA) with sustained receptor binding and optimised physiochemical properties, leading to enhanced tissue penetration [141, 142], an improved side-effect profile and limited drug–drug interactions [143, 144]; it also has no significant inhibitory effects on hepatic bile salt transport [145] and therefore has the potential for a favourable liver safety profile [146]. In the recently published SERAPHIN study, macitentan was shown to be superior to placebo in reducing the number of combined mortality and morbidity events in a large group of patients with PAH [2].
Riociguat is a first-in-class drug that augments cGMP biosynthesis (and therefore vasodilation) through direct stimulation of the enzyme, soluble guanylate cyclase (sGC) in an NO-independent fashion, and by sensitization of sGC to low endogenous NO levels [147]. Results from a multicenter, open-label, uncontrolled phase II trial involving 75 patients with PAH (n = 33) and chronic thromboembolic pulmonary hypertension (n = 42) showed that 12 weeks of oral riociguat given 3 times daily conferred improvements in symptoms, NYHA functional class, exercise capacity, NT-proBNP level, and pulmonary hemodynamics [148]. A decrease in systemic arterial diastolic pressure was the only significant side effect reported, with none associated with symptoms leading to a permanent discontinuation of riociguat [148]. Riociguat is also under investigation in other form of PH such as PH associated with chronic obstructive pulmonary disease, with interstitial lung disease or with left ventricular dysfunction [149–151]. Two large multicentre, randomized, double-blind, placebo-controlled phase III studies have just been published. In PATENT-1, riociguat was shown to be superior to placebo in terms of 6MWT and haemodynamics in patients with PAH [152]. In CHEST-1 riociguat resulted in greater 6MWT and improved haemodynamics in CTEPH patients, either inoperable or with persistent PAH post pulmonary endarterectomy [153].
Future Therapeutic Targets
The following section will cover future potential targets, many of which may target vascular remodelling itself, rather than endothelial cell dysfunction.
PDGF/Tyrosine Kinase Inhibitors
Imatinib, a PDGF/tyrosine kinase receptor inhibitor, was initially shown to be beneficial in a patient with vasodilator-resistant, severe idiopathic PAH [154]. This effect was sustained after 6 months of treatment [154]. A Phase II trial involved 59 patients who were classified as FC II to IV PAH and had an inadequate response to previous therapy were enrolled in a 24 weeks study. The imatinib treated group showed a mean improvement of 22 m in 6MWD compared with a decline of 1 m in the placebo group, although this difference was not significant [155]. However, significant improvements were seen in pulmonary vascular resistance (PVR) (imatinib −300 versus placebo −78 dyn.s.cm-5; p < 0.01) and cardiac output (imatinib +0.6 versus placebo −0.1 L/min-1; p = 0.02). In the recently published Phase III study, IMPRES, imatinib was used as add-on therapy for advanced patients with PAH who were symptomatic despite treatment with two or more PAH-specific therapies. The imatinib treated group showed significantly improvement in exercise capacity and haemodynamics. However, imatinib did not provide a benefit in terms of FC, time to clinical worsening or mortality [156]. There was also a higher incidence of intracranial haemorrhage in the imatanib treated group. As yet it is not known whether further tyrosine kinase receptor inhibitors will have a better efficacy to side-effect profile. Indeed, dasatinib has been shown to induce PAH in certain patients [157].
The EGF receptor tyrosine kinase inhibitors gefitinib, erlotinib and lapatinib have been tested in rats with monocrotaline-induced pulmonary hypertension. These inhibitors have been shown to inhibit EGF-induced smooth muscle cell proliferation in vitro [120]. However, at their highest tolerated dose there was no significant improvement in RV systolic pressure or hypertrophy in animal models. Therefore, more work needed to evaluate the potential of EGFR antagonists as the treatment of PAH.
Epigenetic Modulators
Epigenetics describes changes in phenotype or in gene expression states independent of DNA sequence and include chromatin remodelling, DNA methylation, histone modification and RNA interference. Epigenetic modifications can be inherited or acquired de novo, and provide a mechanism that allows the stable propagation of gene activity states from one generation of cells to the next [158]. It has been shown that epigenetic modifications are involved in many diseases mechanisms including cancer, asthma, several hereditary disorders and recently PAH [159, 160].
DNA Methylation
In the heritable fawn hooded rat pulmonary hypertension model, the expression of superoxide dismutase 2 (SOD2) is decreased and yet no mutations are found in the SOD2 gene [161, 162]. However, CpG islands in the SOD2 gene are selectively hypermethylated, which result in ~50 % reduction of SOD2 expression in pulmonary arterial smooth muscle cells from fawn hooded rats compared to those from genetically matched rats [161].
Histone Methylation and Acetylation
It has been shown that constitutive eNOS expression is increased 6-fold in pulmonary vascular endothelial cells derived from a neonatal rodent perisitent pulmonary hypertension of the newborn (PPHN) model. The eNOS upregulation was associated with increased H3 and H4 histone acetylation in the eNOS promoter [163] and a small decrease in eNOS methylation. Indeed, increased histone deacetylase (HDAC1 and HDAC5) expression and activity has been found in lungs from patients with idiopathic PAH. Treatment with the HDAC inhibitors, valproic acid, and suberoylanilide hydroxamic acid, reduced the constitutive proliferative phenotype of adventitial fibroblasts and rhomboidal cells from hypoxic rat in vitro, and attenuated the development and ameliorated established pulmonary hypertension in a chronic hypoxic rat model [164].
MicroRNAs (miRNAs)
Micro RNAs are small, non-coding RNA molecules that regulate gene expression. miRNAs often target groups of genes that are related in terms of function. There is an increasing literature describing associations of miRNAs with the development of pulmonary hypertension both in in vivo models and in human disease [165–167]. For instance miR 150 is downregulated in the plasma and lungs of patients with idiopathic PAH, and is associated with poorer outcome [168]. Work is currently underway to develop methods to target miRNAs as a novel therapy for PAH and other cardiovascular diseases [169].
Anti-Inflammatory/Immunosuppressive Treatments
There are reports of successful treatment of patients with PAH associated with lupus and mixed connective tissue disease with immunosuppressive agents [170]. Furthermore, a case study reported successful treatment of a patient with Castleman’s-related PAH with the anti-IL-6 monoclonal antibody, tocilizumab [171]. Interestingly, rapamycin, an immunosuppressive agent, has also been investigated in animal models of PAH. Rapamycin is also a potent anti-proliferative agent, and has been shown to be effective in preventing the RV wall thickening in rat monocrotaline model [172]. In addition, rapamycin also inhibited the proliferation of pulmonary vascular cells in vitro and reversed pulmonary vascular remodelling in mice hypoxia-induced pulmonary hypertension model [173]. Finally, the steroid dexamethasone, has been shown to reverse the haemodynamic and structural changes seen in the monocrotaline model of pulmonary hypertension and prednisolone inhibits proliferation of pulmonary artery smooth muscle cells from patients with idiopathic PAH in vitro [174, 175]. Therefore, anti-inflammatory therapy including immunosuppressants steroids, rapamycin and tocilizumab, may provide novel treatments in the future.
Novel Vasoactive Factors
Apelin
Apelin is a vasodilatory peptide which is thought to play a role in angiogenesis and regulate endothelial and smooth muscle cell apoptosis and proliferation [176]. Interestingly, patients with PAH have lower levels of apelin [176]. Currently, clinical trials are testing whether the administrating apelin is beneficial in humans with PAH.
Vasoactive Intestinal Peptide (VIP)
VIP is also a vasodilator [177]. VIP Knockout mice showed significant increases in right ventricular (RV) systolic pressures, vascular remodelling, and inflammation [178], which was attenuated by treatment with VIP. Two clinic trials showed conflicting results. A multicentre Phase II study in 56 patients with PAH suggested that there was no reduction in PVR, or increase in exercise capacity over 12 weeks compared with placebo [179]. The other, single centre, open-label study of eight patients found that haemodynamics and 6MWD were improved following 3 months’ treatment with VIP [180].
Endothelial Nitric Oxide Synthase (eNOS) Couplers
Endothelial nitric oxide synthase can mediate the production of both vasorelaxants and vasoconstrictors: the coupling of eNOS could therefore have a twofold impact on the balance of vasoactive factors released by the endothelial cells [181]. A pilot study of the eNOS coupler, sapropterin dihydrochloride, showed significant improvements in exercise capacity and the treatment was well tolerated, although NO synthesis was not effected [182].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


