Fig. 15.1
Vascular synthesis of cGMP. Cyclic GMP can be synthesised by two distinct mechanisms: NO-stimulated sGC activity and natriuretic peptide-triggered pGC turnover. Therapeutically, sGC-mediated cGMP generation can be recapitulated by NO-donors, inorganic nitrate, and sGC stimulators/activators such as cinaciguat and riociguat. Neutral endopeptidase (NEP) is responsible for the degradation of natriuretic peptides, and inhibition of this enzyme can be used therapeutically to raise endogenous natriuretic peptide bioactivity
Nitric Oxide
Within blood vessels, NO is synthesised in endothelial cells from the substrate l-arginine [66] via an enzyme known as endothelial NO synthase (eNOS, or NOS-3). There are two further isoforms of NOS, both of which are expressed in various vascular and non-vascular locations (see [84]); neuronal (nNOS, NOS-1) and inducible (iNOS, NOS-2) NOS. The production of NO from l-arginine involves a two-step oxidation reaction, utilising both NADPH and molecular oxygen to form l-citrulline and NO [3]. Endothelial NOS, like all NOS isoforms, is a homodimer consisting of a C-terminal reductase domain and an N-terminal oxygenase domain. The reductase domain contains binding sites for NADPH, which donates electrons for the reaction, and FAD/FMN which act as electron conduits. The oxygenase domain contains a heme group and also binding sites for the essential cofactor tetrahydrobiopterin (BH4), and substrate l-arginine. Electrons transfer from NADPH bound at the C-terminal of the reductase domain, to the heme in the oxygenase domain (of the opposite monomer), resulting in the reduction and activation of oxygen, oxidation of l-arginine to l-citrulline and NO generation (reviewed in [19]). In healthy arteries, the major stimulus for eNOS is increased intracellular calcium, which triggers binding of calmodulin to the oxygenase domain, increasing the rate of NADPH-derived electron transfer between the domains. Calcium increases in endothelial cells occur in response to many endothelium-dependent dilators, including bradykinin and substance P. Phosphorylation of eNOS, in response to shear stress, oestrogen, vascular endothelial growth factor, and insulin, also stimulates enzyme activity and NO generation; this is facilitated by non-calmodulin-dependent pathways, such as protein kinase A, Akt and AMP-activated protein kinase [20]. However, in certain cardiovascular diseases, including PH, it is thought that eNOS may exist in an ‘uncoupled state’, in which substrate l-arginine or BH4 become rate-limiting; in this scenario, eNOS transfers electrons to molecular oxygen, rather than oxidising l-arginine, thereby forming superoxide, a species thought to precipitate vascular dysfunction [89]. Thus, it is not only the amount of substrate available for NO production but also the correct coupling of NOS that is important for sufficient NO bioavailability.
Alterations in the synthesis of NO occur in patients with PH and in pre-clinical models of the disease [55]. Endothelial dysfunction is considered to be one of the early events leading to PH, and this is associated with reduced NO synthesis [59]. Interestingly, eNOS expression in the pulmonary vasculature has been shown to decrease in patients with PH [29], whereas other studies report higher levels [95]. These conflicting findings most likely reflect differential mechanisms causing PH, as a third study suggests slow eNOS expression in the pulmonary arterioles of PH patients, but high eNOS levels in the diseased plexiform lesions that occur in pulmonary vessels of PH sufferers [53]. This uncertainty is mirrored in animal models of PH, with a reported increase [48, 71, 85], decrease [77] or no change [59] in eNOS expression shown in various model systems. Furthermore, in eNOS knockout (KO) mice (in which the eNOS gene has been deleted), basal pulmonary resistance is increased [11], but in response to chronic hypoxia, PH disease severity may be worsened [82], unchanged [18] or ameliorated (Johns) compared with wild-type mice. One explanation for these differences in eNOS expression found in PH patients and disease models is the uncoupling of the enzyme (as mentioned above). Thus, whilst lower enzyme expression and activity (i.e. NO production) can contribute to pulmonary vasoconstriction, higher levels of eNOS protein may not necessarily be beneficial if uncoupling of the enzyme leads to superoxide production (rather than NO) and thereby exacerbates the disease. This thesis is supported in hph1 mice, which have an inherent BH4 deficiency. Exaggerated responses to hypoxia-induced PH as a result of eNOS uncoupling are found in these animals [64]. A recent report has also highlighted a novel mechanism whereby eNOS-mediated oxidative stress impairs PKG phosphorylation [102]. Under normal conditions eNOS is inhibited by coupling to caveolin-1, however when caveolin-1 expression is decreased eNOS can become abnormally hyperactive and influence superoxide production. Thus, the level of eNOS activity per se is not necessarily as important as the products and downstream signalling that it evokes.
NO-Based Therapeutics
The balance between eNOS-driven NO production and enzyme uncoupling has been targeted as a potential therapeutic avenue for PH. Supplementation with the eNOS cofactor, BH4, or a more stable analogue, has been investigated and can effectively normalise eNOS expression and endothelial function [46]; such an approach may prove to be effective in PH [4]. However, BH4 supplementation for 4 weeks prior to coronary artery bypass did not improve endothelial function in patients with coronary artery disease, thought to be because of oxidation of BH4 to BH2 [14]. This oxidation may have been due to the oral administration of the drug, thus, modification of the route of administration may still uncover an effective treatment. Another drug, cicletanine, which is presumed to work by coupling eNOS/BH4 activity to promote NO (rather than superoxide) production is beneficial in animal models of PH [36] and decreases pulmonary resistance in PH patients [73], although these effects may also be attributed to natriuretic peptides or cAMP. Unfortunately, clinical trials did not find cicletanine to be effective in the treatment of PAH and further development of the drug for this indication is unlikely.
In some cases, NO production may be limited by the substrate, l-arginine, and increasing this may be useful in enhancing NO synthesis. l-arginine administration attenuates PH in both hypoxic and monocrotaline-treated rats [57] and acutely decreases pulmonary vascular resistance in PH patients [54]. Short-term administration of oral l-arginine has been reported to improve pulmonary hemodynamics in patients with PH [58, 63]. However, no extended clinical trials have been undertaken in patients with PAH and the effectiveness of this approach is yet to be determined.
In general, PH is associated with reduced bioavailability of NO. Thus, attempts have been made to simply increase its level in the pulmonary circulation by having patients inhale it. Inhalation of just trace amounts of NO, in the range of 1–20 parts per million (ppm) have been shown to have an acute pulmonary vasodilator effect. Inhaled NO has been developed and approved for treatment of infants with persistent pulmonary hypertension of the newborn and is presently undergoing clinical trial in adult patients with PAH.
Inhaled NO has the advantage of being delivered directly to pulmonary vessels and thus largely avoids the systemic vasodilation associated with other modes of delivery [81]. This method of delivery also results in better ventilation/perfusion (V/Q) matching since more NO is delivered to better ventilated areas of the lung. However, the use of inhaled NO to treat patients with PAH has been hampered by its extremely short half-life resulting in the need for near continuous inhalation and uncertainty regarding potential for pulmonary hypertensive rebound response if inhalation is abruptly interrupted. In addition, inhaled NO therapy is plagued by the need for a cumbersome and expensive delivery device [4, 55, 81]. In an attempt to bypass some of these problems, NO donors have been developed that provide longer-lasting NO release. Nebulised NONOates [31, 87] and inhaled glycerol trinitrate [30] are both effective in reducing pulmonary vascular resistance; however, there are still concerns about the lack of pulmonary specificity with these drugs [4]. An alternative ‘NO-donor’ therapy may be to enhance endogenous NO production by administering inorganic nitrite and/or nitrate. A study from our own laboratory has shown that in animal models of PH, induced by either chronic hypoxia or secondary to bleomycin-induced lung fibrosis, several disease parameters are reversed by oral therapy with either of these compounds [5]. This strategy relies on a recently discovered phenomenon where inorganic nitrite is converted to NO and O2 by a variety of heme-based enzymes including haemoglobin and myoglobin especially under conditions of hypoxia and acidosis [37, 86]. The advantage of this approach is that production of NO is maximised in the lung in models of PH, suggesting it should represent a more pulmonary-specific strategy. Moreover, inorganic nitrates can be derived from natural dietary sources such as beetroot and green leafy vegetables, intimating this may be a cost-effective treatment regimen. Clinical evaluation of inorganic nitrite and nitrate administration in PH patients is on-going.
Soluble Guanylate Cyclase
Soluble GC is a heterodimer formed of alpha and beta subunits, and both subunits are required for catalytic activity. In addition, a prosthetic heme group is essential for sGC activation by NO, as NO-triggered enzyme turnover is abolished when the heme group is removed and restored when the heme group is reconstituted (for review see [22]). Activation of sGC by NO involves the uncharged NO radical binding a reduced Fe2+ heme moiety. NO binding to this prosthetic group results in a conformational change which activates the enzyme, increasing the V max more than 200-fold. Due to this heme-dependency for NO-mediated activation, changes to the redox state of sGC, which may occur in cardiovascular disease, including PH, can result in down-regulation of activity and/or expression [69]. Importantly, from a therapeutic perspective, sGC can be activated by synthetic compounds even when it is in an inactive state. In PH the expression and activity of sGC is up-regulated, probably as a compensatory mechanism to adapt to reduced NO bioavailability [4] and sGC-null mice showed an exaggerated response to hypoxia-induced PH [90]. Therefore, sGC has proven to be an important candidate target for PH therapy.
Soluble GC activators, such as cinaciguat (BAY 58-2667), can stimulate sGC when it is in an oxidised or heme-free state, whereas sGC stimulators (e.g. riociguat; BAY 63-2521) can induce sGC activation and stabilise the enzyme in its active configuration via allosteric binding. Thus, sGC stimulators have synergistic effects with NO because they act directly on sGC to induce cGMP synthesis while enhancing the ability of sGC to respond to NO. For example, BAY 41-2272 alone increases sGC activity by ~20-fold but in combination with subthreshold concentrations of NO, sGC activity is increased 200-fold [80]. In comparison, sGC activators only cause additive effects when combined with NO. In rodent models of PH, treatment with either sGC activators such as cinaciguat or sGC stimulators, such as BAY 41-2272 [15] or riociguat [47] attenuates disease severity. The reliance on an intact NO generating system is evident as treatment effects of BAY 41-2272 are abolished in eNOS KO mice [15]. The sGC stimulators have also been shown to attenuate PH in a wide range of other animal studies (summarised in [80]).
The efficacy of sGC stimulation observed in animal models of PH led to the development of riociguat for the treatment of PAH [26]. Following the successful completion of a phase II trial in patients with PAH, 2 large randomised, placebo-controlled studies were conducted to evaluate the efficacy of riociguat for treatment of PAH and chronic thromboembolic disease [27, 28]. In both studies, riociguat improved 6-min walk distance, pulmonary hemodynamics and delayed time to clinical worsening (Fig. 15.2a, b). These studies led to the approval of riociguat for treatment of PAH and CTEPH in 2013.
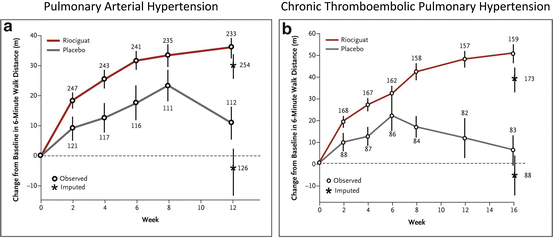
Fig. 15.2
Effect of soluble guanylate cyclase stimulator on 6-min walking distance (a) Mean (±SE) changes from baseline in the 6-min walk distance over 12 weeks in the PATENT-1 study where patients with pulmonary arterial hypertension received riociguat at a dose up to 2.5 mg or placebo three times daily (From [27]). (b) Mean (±SE) changes from baseline in the 6-min walk distance over 16 weeks in the CHEST study where patients with chronic thromboembolic pulmonary hypertension were treated with riociguat or placebo (From [28]). For both (a) and (b), the number at each data point indicates the number of patients included in the assessment at that time point
Natriuretic Peptides
There are three structurally similar vasoactive members of the natriuretic peptide family: atrial, brain and C-type natriuretic peptide (ANP, BNP, CNP). These peptides are generated after enzymatic cleavage of precursor pre-propeptides facilitated by the endopeptidases, corin, in the case of ANP/BNP [96], and furin for CNP [94]. ANP and BNP act in an endocrine fashion and are primarily released in response to hypervolemia. These hormones, as their names suggest, are responsible for regulating salt (natriuresis) and fluid (diuresis) homeostasis. ANP and BNP are stored in granules in the cardiac atria and released in response to atrial wall stretch in response to increased intravascular volume and are then distributed to target tissues such as the kidneys and systemic vasculature. BNP is also synthesised and released from the cardiac ventricles (the name refers to its original site of identification) particularly in pathological situations. The plasma levels of ANP and BNP rise sharply in patients with congestive heart failure (CHF) and PH, and correlate with disease severity and survival. Thus, these peptides are often used as biomarkers; this is particularly true of BNP or its N-terminal inactive fragment, NT-proBNP [62, 67].
A third member of the natriuretic peptide family, CNP, is highly expressed in the brain, chondrocytes and vascular endothelial cells and is secreted from the latter cell type at an increased rate in response to inflammation and shear stress.
The natriuretic peptides increase intracellular cGMP levels by binding to cell surface GC-linked receptors, commonly referred to as particular guanylate cyclases (pGC) (Fig. 15.1). The natriuretic peptide receptors A and B (NPR-A and NPR-B) are transmembrane receptors which contain a ligand-binding extracellular domain, an intracellular protein kinase-like domain and a C-terminal GC catalytic region. The protein kinase like domain binds ATP, which leads to increased NP-induced GC stimulation. Conversely, the receptor can become desensitised by dephosphorylation at this region [56]. NPR-A is highly expressed in renal, vascular smooth muscle, heart and lung tissue and is most sensitive to ANP, followed by BNP and, at high (pharmacological) concentrations, CNP [67]. Evidence of NPR-A being solely responsible for the natriuretic/diuretic actions of ANP has been shown in NPR-A knockout mice, where these mechanisms were completely lost [40]. NPR-B is the major receptor for CNP, and essential for normal bone growth (loss-of-function mutants of NPR-B have severe bone deformities and dwarfism) [10]. A third NPR, NPR-C, is known as the natriuretic peptide clearance receptor. This protein is particularly abundant in the vasculature; indeed it is the most highly expressed of the NPRs, but it is not linked to a GC domain. As its name suggests, NPR-C plays an important role in removing all three NPs from the circulation. Upon binding to NPR-C, natriuretic peptides are rapidly internalised within the cell and degradation of the peptides occur by lysosomes, before the receptor is returned to the cell surface [51]. Under normal circumstances, the affinity of all three natriuretic peptides for NPR-C is similar; however, a pathophysiological environment can result in alterations in NPR-C affinity and expression. As an example, under conditions of acute or chronic hypoxia, NPR-C expression can be down-regulated [41, 49], presumably as a compensatory response to maintain higher levels of protective natriuretic peptides.
The physiological effects of increasing concentrations of ANP or BNP include direct vasodilatation due to cGMP-mediated smooth muscle relaxation, increased glomerular filtration, reduced salt and water reabsorption, lower renin secretion and aldosterone synthesis, all of which reduce blood volume, and subsequently blood pressure. ANP and BNP also have direct effects on the heart, and are involved in reducing hypertrophic growth (ANP) and fibrosis (BNP). Importantly, ANP and BNP have been shown to blunt the development of pulmonary hypertension in animals and mice lacking NPR-A have increased pulmonary vascular remodelling and right ventricular mass and develop more severe pulmonary hypertension when exposed to chronic hypoxia [45, 99]. These findings suggest that ANP and/or BNP are important in the regulation of pulmonary vascular tone and right heart hypertrophy.
The majority of the effects of CNP are mediated via a different pGC, NPR-B. Ligand binding to NPR-B induces cGMP synthesis causing local vasodilation, rather than an endocrine effect on blood volume and pressure. However, recent evidence also suggests cGMP-independent vasodilation by CNP which is mediated via NPR-C (a non-guanylate cyclase-coupled receptor) [2]. Whether CNP has a role in PH remains uncertain. Infusion of exogenous CNP does not show efficacy in two well characterised models of PH; either chronic hypoxia alone or in combination with SU5416 (a vascular endothelial growth factor receptor inhibitor). However, exogenous CNP was shown to reduce monocrotaline-induced PH and increase survival [34]. Further work is required to establish any function of endogenous CNP in the pathogenesis of PH.
Natriuretic Peptide-Based Therapy
Exogenously administered natriuretic peptides have shown promise as treatments for PH. Treatment with ANP [35] and BNP [44] attenuates right ventricular hypertrophy and pulmonary vascular remodelling in rats with hypoxia-induced PH. In humans exposed to acute hypoxemia, BNP infusion significantly reduced pulmonary vascular resistance compared with placebo [9]; whilst ANP was not effective in this study, in a small cohort of patients with PH secondary to chronic obstructive pulmonary disease, ANP infusion lowered pulmonary vascular resistance by a third and dose-dependently reduced pulmonary artery pressure [1]. BNP was also found to lower mPAP in patients with PAH when combined with a phosphodiesterase inhibitor [43]. Thus, ANP and or BNP might be considered treatment options for PH. However, these peptides have short plasma half-lives (e.g. ANP has a half-life of <1 min in rats [61] and <3 min in humans [97]) and they are not amenable to oral administration, making them poor candidates. An alternative strategy is to enhance endogenously produced natriuretic peptides by preventing their clearance or degradation. As NPR-C is involved in the clearance of natriuretic peptides, blockade of NPR-C can increase the bioavailability and therefore the biological effects of ANP and BNP. Thus, antagonists at this receptor, which have been developed [88] could increase bioavailability of natriuretic peptides, making this a useful therapeutic target [5]. A second mechanism by which bioavailability of natriuretic peptides can be enhanced is by inhibiting their degradation. Neutral endopeptidase (NEP) metabolises natriuretic peptides by cleavage and inactivation; thus NEP inhibitors, such as ecadotril, can boost endogenous natriuretic peptides. NEPs are not selective for natriuretic peptides, they metabolise a vast array of peptides, many of which are involved in regulation of vascular tone, including angiotensin II, bradykinin, endothelin-1 and substance P. Thus, the use of NEP inhibitors to enhance natriuretic peptides may be offset by enhancement of vasoconstrictor peptides (for review see [52]). Nevertheless, the relative specificity of natriuretic peptides for the pulmonary circulation results in a beneficial effect of NEP inhibition for the treatment of PH [6, 42]. Moreover, inhibition of NEP appears to specifically enhance natriuretic peptide activity in the pulmonary circulation, entailing a relatively pulmonary-selective activity.
cGMP Metabolism
The balance between the synthesis of cGMP (discussed above) and the breakdown by phosphodiesterase enzymes largely determines the dynamic, local concentrations of cGMP available for downstream signalling. There is also a role for ATP-dependent multidrug resistance transporter proteins in exporting cGMP from smooth muscle cells; however, this effect is reported to be small compared to the role of PDEs in regulation of cGMP [21]. Nonetheless, pharmacological inhibition or genetic deletion of MRP activity has been shown to ameliorate experimental PH. Figure 15.3 shows an overview of cGMP signalling in the vasculature, including the role of PDE isozymes.
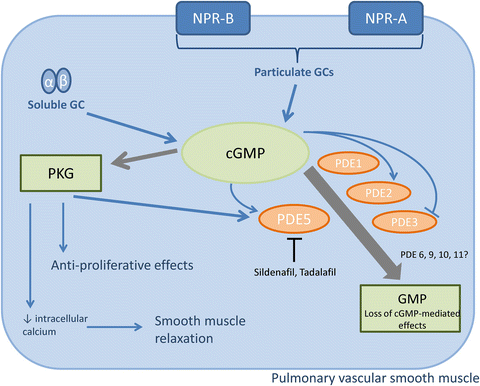
Fig. 15.3
Metabolism and action of vascular-derived cGMP. The major actions of cGMP in vascular smooth muscle are relaxation and anti-proliferation, mediated by protein kinase G (PKG). The bioavailability of cGMP can be reduced by phosphodiesterase (PDE)-mediated hydrolysis. Within vascular tissue, PDE5 is the best characterised cGMP-specific PDE, and is the primary target to enhance cGMP bioavailability therapeutically. Sildenafil and tadalafil are PDE5 inhibitors licensed for use in PH. Other PDEs that hydrolyse cGMP may be involved in pulmonary vascular hemodynamics and the pathogenesis of PH
PDE-Dependent Hydrolysis of cGMP
Cyclic GMP is degraded to inactive GMP by PDEs thereby reducing the substrate available for PKG activation. There are 11 known families of PDEs [1–12], each of which contain 1–4 gene-related isozymes/splice variants, encoding, in total, more than 100 different PDE proteins [39]. These enzymes can metabolise either cGMP, cAMP or both. PDEs 4, 7 and 8 have no, or minimal, cGMP hydrolysing activity and are not discussed further in this chapter. Of the remaining PDEs, all hydrolyse cGMP to varying degrees. To date, PDE5 is the most well characterised cGMP metabolising PDE and is thought to be the most active cGMP hydrolysing PDE in smooth muscle under basal conditions [72].
PDE5 is a homodimer, with both monomers containing a regulatory and a catalytic domain. The regulatory domain contains two GAF cGMP binding sites, GAF-A and GAF-B. PDE5 binds to cGMP at a high affinity at the GAF-A binding site. Cyclic AMP can also bind to this GAF site, however PDE5 is ~100-fold less selective for cAMP over cGMP. Upon binding, catalytic activity is enhanced whereas when this GAF-A binding site is blocked, inhibition of enzyme activity occurs. Inhibitors of PDE5 that are currently in use act by inhibiting the catalytic domain. Adjacent to the GAF-A binding site is a serine residue which is phosphorylated by PKG. This forms a negative feedback loop, whereby cGMP-stimulated PKG activity inhibits further cGMP signalling (reviewed in [38]). Expression of a single gene, PDE5A, accounts for the biological activity attributed to PDE5, although this gene has three splice variants, PDE5A1, PDE5A2 and PDE5A3. This gene is widely expressed, with relatively high levels of PDE5A found in platelets, kidneys, cerebellum, and pancreas, but it is most abundant in vascular smooth muscle and lungs [12]. PDE5 expression is relatively high in the pulmonary circulation, and indeed is up-regulated in the pulmonary circulation of both animals and patients with PH [60, 75, 76, 92]. PDE5A is also up-regulated in the right ventricle after pressure-overload hypertrophy.
PDE5 Inhibition for Pharmacotherapy in PH
Over recent decades, the therapeutic potential of PDE5 inhibitors for treatment of PH has been vigorously investigated. The PDE5 inhibitor, sildenafil, was initially trialled as anti-angina medication, and it was by serendipitous observations that the beneficial effect on penile erection was discovered, which led to the widespread use of sildenafil, marketed as Viagra, for the treated of erectile dysfunction. It was found that PDE5 expression was particularly high in the smooth muscle cells of the corpus cavernosum of the penis, which explained why smooth muscle relaxation, which is required to increase blood volume within the penis, was stimulated at this site [70].
After this discovery, the use of sildenafil in other regions with high PDE5 expression was considered. Following promising results from several small clinical trials, a large randomised placebo-controlled trial of sildenafil for treatment of PAH was conducted [25]. Sildenafil was given at a dose of 20, 40 or 80 mg three times daily. After 12 weeks of treatment, 6-min walking distance and pulmonary hemodynamics were significantly better in patients in any of the sildenafil treatment groups compared to placebo (Fig. 15.4a). Sildenafil was subsequently approved at the lowest of the three doses (20 mg three times daily) for the treatment of PAH in 2005.
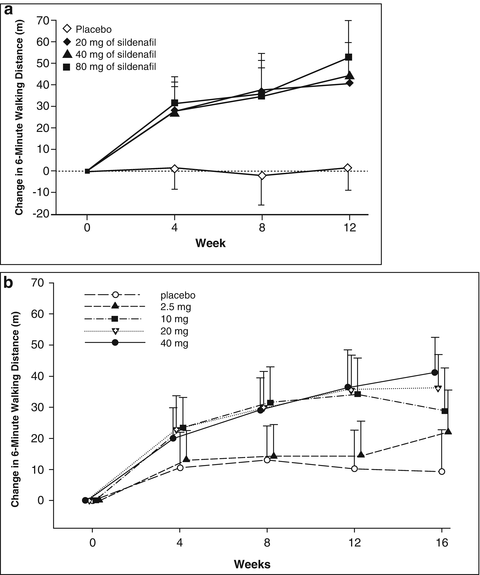
Fig. 15.4
Effect of phosphodiesterase-5 inhibitors on 6-min walking distance (a) Mean changes from baseline, with 95 % confidence intervals, in the 6-min walking distance over 12 weeks in the SUPER study where patients received sildenafil of placebo three times daily (From [25]). (b) Mean changes from baseline, with 95 % CIs, in the 6-min walking distance over 16 weeks in the PHIRST study where patient received tadalafil or placebo once daily (From [23])
Adult patients with PAH have similar acute pulmonary vasodilator responses to any of the three presently available PDE5 inhibitors that are presently available to treat erectile dysfunction. In addition to sildenafil, vardenafil and tadalafil have been shown to improve PAH in adults. Vardenafil is not approved for the treatment of PAH in the US, but tadalafil was approved for this indication in the US in 2009 following the completion of a randomised controlled trial of 2.5, 10, 20 or 40 mg tadalafil given once daily compared to placebo [23]. In that study, only the 40 mg dose was successful as defined a priori as improvement in placebo-adjusted 6-min walk test after 16 weeks of treatment at pre-specified significance level of P > 0.01 (Fig. 15.4b). Of the PDE5 inhibitors licensed for use in PH, tadalafil is longer lasting and more specific to PDE5 relative to PDE1-4 than sildenafil [39]. Importantly, in humans with PH both sildenafil [25] and tadalafil [23] are effective in improving symptoms and outcomes.
Pre-clinical studies have provided insight into the therapeutic mechanisms of PDE5 inhibitors. It was established that sildenafil is effective in inhibiting PH in chronically hypoxic mice [100], in monocrotaline-treated rats [74], and in acutely hypoxic neonatal lambs [91] and pigs [78]. In fact, a plethora of experimental evidence exists supporting the thesis that PDE5 inhibition can improve PH in animal models [81]. However, as with patient responses to sildenafil (~45 % patients do not respond favourably), there is still significant room for improvement in treatment options. Therefore investigators, both pre-clinical and clinical, are now focussing on combination therapies. It is clear that targeting one element of cGMP signalling alone may not substantially increase cGMP, or at least be maximally efficacious. For example, reducing the degradation of cGMP may be ineffective if there is not sufficient substrate for cGMP generation. Thus, an improved efficacy may be apparent by employing a strategy based on increasing cGMP production at the same time as reducing degradation. In this regard, mice lacking NPR-A are less responsive to chronic sildenafil treatment in models of PH [101] suggesting that natriuretic peptides are essential for PDE5 inhibition to succeed. In accord, the beneficial effects of sildenafil to reduce right ventricular pressure in rats exposed to acute hypoxia are augmented by concomitant infusion of ANP [68]. Further still, treatment with the NEP inhibitor ecadotril (to enhance endogenous natriuretic peptides) and sildenafil results in a synergistic reduction in right ventricular pressure and hypertrophy in chronically pulmonary hypertensive rats [6]. Further still, in humans, BNP alone does not reduce pulmonary artery pressure acutely, but enhances the benefits of sildenafil [43]. Together, the studies described above demonstrate that the generation of cGMP due to increased natriuretic peptide bioactivity underpins the benefit of PDE5 inhibition in PH. In accord, it is therefore conceivable that NO therapy would produce a similar result. Thus, it has recently been shown that combined treatment with an sGC stimulator (BAY 41-8453) and a PDE5 inhibitor (zaprinast) results in prolonged pulmonary vasodilation in a model of PH [17]. In addition, in response to chronic hypoxia, sildenafil treatment is more effective in reducing PH in wild-type compared with eNOS knockout mice [100]. However, the use of PDE5 inhibitors in combination with nitrovasodilators is contraindicated (due to the development of life-threatening systemic hypotension), due to a lack of pulmonary specificity. In addition, combined use of riociguat and sildenafil or tadalafil is contra-indicated due to risk of symptomatic hypotension [24]. Therefore, combination therapy targeting natriuretic peptide-stimulated cGMP is likely to offer a superior, pulmonary-specific approach.
PDE1
PDE1 is thought to become the most predominant in hydrolysing cGMP under conditions of high calcium, such as occurs during smooth muscle contraction [72]. Unlike PDE5, PDE1 is expressed at low levels in the pulmonary tissues. However, PDE1 is substantially up-regulated in pulmonary vasculature undergoing proliferation. In both patients with PH and in monocrotaline-treated rats, PDE1 expression is greater in pulmonary arteries and smooth muscle cells compared with basal expression. Furthermore, inhibition of PDE1 reduces right ventricular pressure and pulmonary resistance [75]. PH induced by chronic exposure to cold temperature in rats is also reversed by PDE1 inhibition [13]. Likewise, PDE1 inhibition enhances the vasodilator response to NO in an acute hypoxia PH model in lambs [16]. These data advocate PDE1 as a therapeutic target for PH.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


