Fig. 3.1
Schematic diagram of human mitochondrial genome to show the mtDNA-derived 13 polypeptides, 22 tRNAs (each was named based on respective amino acid), and two rRNAs (12S and 16S). It also shows the reported mutations/polymorphisms in asthmatics along with respective associated asthma phenotypes
In support of a direct role for mitochondria in asthma, common mitochondrial haplogroups, especially haplogroup U from European population, have shown association with increased serum IgE levels [35]. Another study found five mutations in mitochondrial gene-encoding tRNAs (A12308G in tRNA leucine (CUN), G15928A in tRNA threonine, 595insC in tRNA phenylalanine, T10448C in tRNA arginine, and A8343G in tRNA lysine) and five more mutations in tRNA flanking regions [37]. Importantly, A12308G mutation is part of the mitochondrial haplogroup U, which was found to be significantly associated with total serum IgE levels. Another case-control study in Caucasian European children has found an association of the genomic region containing the ATPAF1 (ATP synthase mitochondrial F1 complex assembly factor 1) gene in Caucasian European children [38]. Further, they found that ATPAF1 gene expression was increased in asthmatic bronchial biopsies [38]. In other studies, peripheral blood mononuclear cells (PBMCs) of allergic patients were found to differentially express nine mitochondrial genes, including II and III subunits of cytochrome c oxidase [39]; cytochrome b gene polymorphism was linked with nonallergic bronchial asthma [40]; and substitution polymorphism at 195 site and insertion polymorphism at 309 site of hypervariable regions of D loop of mitochondria were associated with bronchitis in children [41]. Interestingly, A3243G tRNALeu(UUR) mutation which is associated with MIDD (maternally inherited diabetes and deafness syndrome) and MERRF (myoclonic epilepsy and ragged-red fibers) syndromes has been found in rare forms of asthma along with other comorbidity conditions such as maculopathy or depression [42, 43]. It is to be noted that A3243G tRNALeu(UUR) leads to severe combined respiratory chain assembly defect with abnormal incorporation of amino acids, alteration in stabilization, methylation, aminoacylation, and codon recognition of tRNA [44]. These indicate the necessity of initiating a study of mitochondrial variome in various populations to understand the exact role of mitochondria in the pathogenesis of asthma and other diseases.
Mitochondrial Function and Asthma
It is also necessary to note that mitochondrial research has seen periodic surges related to discoveries of new functions of mitochondria, starting from initial descriptions as energy sources to critical orchestrators of cell death [45]. Meanwhile, the understanding and management of asthma have also seen various evolutions (from the concept of smooth muscle defect to airway remodeling), thanks to the rapid advances in the field of immunology and molecular biology [46, 47]. While a link between the two was not realized until recently, many common features can be seen between mitochondrial pathobiology and asthma pathophysiology (Fig. 3.2).
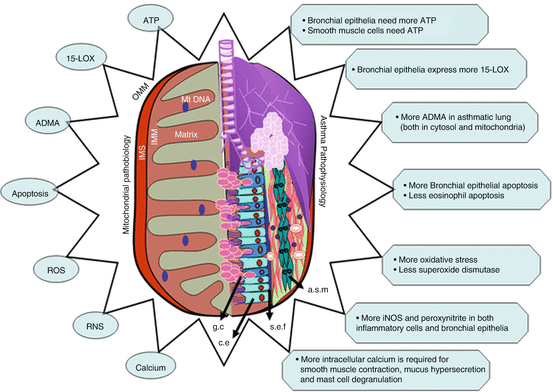
Fig. 3.2
Schematic diagram shows the respiratory organelle of the cell on one side and respiratory organ of body on the other side to indicate the possible common aspects between mitochondrial biology and asthma pathogenesis. These are ATP generation, 15-lipoxygensase (15–LOX), asymmetric dimethyl L-arginine (ADMA), apoptosis, reactive oxygen free radicals (ROS), reactive nitrogen free radicals (RNS), and calcium homeostasis. OMM outer mitochondrial membrane, IMS intermembranous space, IMM inner mitochondrial membrane, g.c. goblet cell, c.e. ciliated epithelia, s.e.f. subepithelial fibrosis, a.s.m. airway smooth muscle
Mitochondrial Dysfunction
Before our lab demonstrated the potential role of mitochondrial dysfunction in asthma, there were scattered reports indicating increased number of mitochondria in the bronchial epithelia of asthmatic children and experimental allergic mice [17, 18]. The functional status of such mitochondria was not known. We found that allergic murine lungs show features of mitochondrial dysfunction like decreased cytochrome c oxidase activity in lung mitochondria, decrease in the expressions of the third subunit of cytochrome c oxidase and 17 kDa subunit of complex I in bronchial epithelium, decreased lung ATP levels, and increased cytochrome c in lung cytosol [19]. Importantly, this was associated with mitochondrial ultrastructural changes such as mitochondrial swelling and loss of cristae. Further, IL-4 monoclonal antibody administration reduced these mitochondrial dysfunction and ultrastructural changes, indicating that mitochondrial dysfunction is associated with asthma pathogenesis (Fig. 3.3) [19]. In another study, mice exposed to environmental irritants such as tobacco smoke and printer emissions exhibited damage to lung mitochondria along with increased generation of reactive oxygen species (ROS) [48]. However, whether the observed mitochondrial dysfunction was the cause or effect in asthma pathogenesis remained uncertain. Findings from Boldogh lab shed some light in this context, where they demonstrated that increase in ROS generation from mitochondrial respiratory complex III augments Th2 responses independent of the adaptive immune response [49]. Further, they demonstrated that preexisting mitochondrial dysfunction in airway epithelia which was induced by deficiency of UQCRC2, ubiquinol-cytochrome c reductase core II protein, worsened asthma features in ragweed pollen-induced allergic mice [20]. These data indicate that mitochondrial dysfunction may be an important component of asthma pathogenesis and that restoration of normal mitochondrial function may be an important target for improving asthma. In support of these findings from mouse studies, dysfunctional mitochondria have also been observed in human asthmatic bronchial epithelia [50].
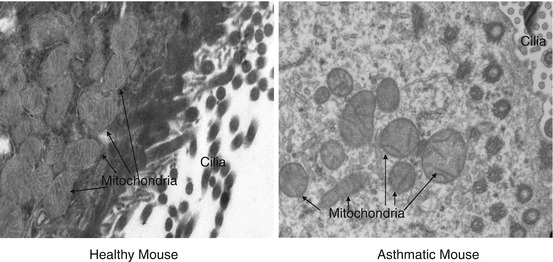
Fig. 3.3
Transmission electron microscopy of bronchial epithelia of asthmatic mice
Reactive Oxygen Species
It is a well-known fact that mitochondria-mediated aerobic metabolism is, bioenergetically, at least 19 times more efficient than anaerobic glycolysis [51]. However, this energetically favorable system also introduces oxidative free radicals as unwanted by-products. Mitochondria are the major endogenous source of oxidative free radicals, and oxidative stress is tightly linked with airway inflammation. Additionally, asthmatic airway is bombarded with various proinflammatory mediators from various recruited inflammatory cells such as eosinophil, lymphocytes, macrophages, neutrophil, mast cells, etc. This leads to increased oxidative stress in the lung. For example, eosinophil secretes various toxic proteins such as eosinophil peroxidase, major basic protein, eosinophil cationic protein, and eosinophil-derived neurotoxin [52]. These various proinflammatory mediators create local oxidative milieu in the airway [53]. The fate of generated superoxide (O2 −) or free radicals is largely dependent on the local microenvironment. If there are sufficient dismutase enzymes inside the mitochondria, superoxide (O2 −) is converted to H2O2, which is relatively stable and can be further broken down by catalase. However, superoxide dismutase has been shown to be reduced in asthmatic airway [54, 55]. Under these conditions, unstable superoxide may directly damage the mitochondria. This is consistent with the reports of mitochondrial dysfunction in asthma [19, 20].
The molecular targets of mitochondria-generated superoxide (O2 −) are similar to any other parts of the cell such as nucleic acids, proteins, and lipids, similar to the NADPH oxidase-derived ROS. However, since mitochondria lack sophisticated DNA repair mechanisms, mitochondrial DNA mutations can accumulate [56]. Mitochondrial damage due to accumulated mitochondrial DNA mutations is an attractive theory of aging [57]. However, whether the reduction in the mitochondrial function due to accumulated mutations leads to aggravation of existing asthma features or induction of new asthma is yet to be investigated. To support this, various studies have found association of mitochondrial gene variations with asthma [35–42].
Oxidative damage to mitochondrial lipids such as cardiolipin, which is vulnerable due to its unsaturated state, leads to inhibition of the respiratory complexes [58, 59]. Oxidative stress also induces prooxidant enzymes such as 15-lipoxygenase (15-LOX), a lipid-peroxidizing enzyme, which is present in cytosol [60]. 15-LOX is unique since it directly hydrolyzes phospholipid esters present in biomembranes such as mitochondrial membrane even without the prior action of phospholipase A2 (PLA2) [60]. A number of evidences show that 15-LOX may be the common denominator between asthma pathogenesis and mitochondrial dysfunction. 15-LOX is the main enzyme in degrading mitochondria in reticulocyte during RBC maturation, attacks mitochondria under oxidative stress conditions, and can lead to mitochondria-mediated apoptosis [59]. In the lungs, 15-LOX is inducible by important Th2 cytokines such as IL-4 and IL-13, which are sufficient to induce an asthma-like state. Further, 15-LOX is predominantly expressed in the bronchial epithelium, where mitochondrial dysfunction and apoptosis have been noted [60]. In mice, deficiency of 12/15-LOX has been shown to alleviate the features of asthma [61, 62]. Together, this suggests that IL-4- or IL-13-mediated increase in 15-LOX may cause epithelial mitochondrial dysfunction and injury, which in turn lead to asthma. However, neither the exact role of 15-LOX in mitochondrial degeneration nor the consequences of epithelial mitochondrial dysfunction that leads towards asthma pathogenesis are well understood.
Reactive Nitrogen Species
Nitric oxide synthase (NOS) generates NO from substrate L-arginine, a semi-essential amino acid, and there are different isoforms of NOS: constitutive and inducible [63]. The role of NO in asthma is controversial and depends on its cellular source, isoform, and microenvironment [63, 64]. In normal airways, NO from constitutively expressed NOS such as eNOS (endothelial NOS) reduces airway tone by bronchodilation via cGMP pathway and increases mitochondrial biogenesis [63, 64]. In asthmatic airways, high levels of arginase consume most of the endogenous L-arginine and thus lead to reduced substrate availability to eNOS, inhibiting its bronchodilator action. Increased expression of iNOS during inflammation along with high levels of ROS leads to formation of reactive nitrogen species, peroxynitrite, which can cause numerous cytotoxic activities including mitochondrial dysfunction and is also a potent bronchoconstrictor [63]. NO and peroxynitrite inhibit cytochrome c oxidase (COX), a key oxidative enzyme of mitochondrial electron transport chain [65, 66]. Peroxynitrite not only affects cytochrome c oxidase but also affects many other mitochondrial enzymes such as NADH/coenzyme-Q reductase, aconitase, cytochrome c reductase, ATP synthase, and succinate dehydrogenase [67]. Thus, peroxynitrite is more effective to cause mitochondrial damage compared to NO. Thus, NO has a dual role in asthma pathophysiology as well as mitochondrial biology. In this context, therapeutic strategies for increasing eNOS and/or decreasing iNOS and peroxynitrite would be beneficial for both asthma and mitochondria. In this context, a high dose of L-arginine (250 mg/kg) reduces the formation of peroxynitrite by regulating NO and asymmetric dimethylarginine (ADMA) metabolism and alleviates the asthma features along with the reduction in mitochondrial dysfunction and epithelial damage [64, 68–70]. Since the existence of mitochondrial NOS is still under debate, we are not discussing that here [71].
Increased peroxynitrite formation in asthma can also be explained by the high levels of asymmetric dimethyl arginine (ADMA). ADMA binds to NOS, displacing L-arginine, and cannot be oxidized to NO, instead leading to formation of superoxide by uncoupling electron flow and NO production [69]. Although it has been suggested that ADMA can inhibit all forms of NOS, it predominantly inhibits eNOS in airway epithelia compared to inducible NOS [63, 68, 69]. The uncoupling of NOS leads to production of superoxide and peroxynitrite, which leads to mitochondrial dysfunction in pulmonary arterial endothelial cells [72]; however, the exact role of ADMA in asthma is yet to be explored. We have shown increased ADMA levels, along with increased expression of ADMA-synthesizing enzyme and decrease in the expression of ADMA-degrading enzyme, in bronchial epithelia of asthmatic lungs [69]. Importantly, ADMA levels were found to be increased in lung mitochondria in asthmatic mice [69]. Also, ADMA infusion leads to increased collagen deposition and deterioration of lung function in mice [73]. Further, ADMA increases the levels of both superoxide free radicals and peroxynitrite in bronchial epithelia (Fig. 3.4) [74].
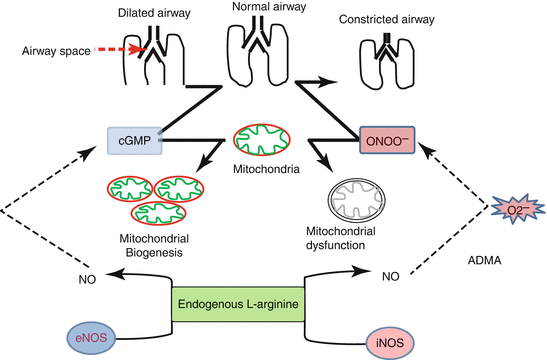
Fig. 3.4
Schematic diagram to show the dual role of nitric oxide (NO) in bronchial asthma and mitochondrial biology. Endogenous L-arginine is the common substrate for both eNOS (endothelial NOS) and iNOS (inducible NOS). NO derived from eNOS activates cGMP which dilate the airway; on the other hand cGMP also leads to mitochondrial biogenesis, whereas NO derived from iNOS reacts with superoxide free radicals to form peroxynitrite (ONOO−) which is the powerful bronchoconstrictor and also causes mitochondrial dysfunction. Increased ADMA leads to increase in the formation of superoxide free radicals and peroxynitrite
Apoptosis
Epithelial injury and shedding are increased in asthma. This may relate to increased mitochondria-mediated apoptosis in bronchial epithelia as oxidative stress in the lung could lead to cardiolipin peroxidation that dissociate cytochrome c from cardiolipin and release into cytosol where cytochrome c complexes with Apaf-1 and deoxy-ATP to form apoptosome which further activates caspase 9 and caspase 3 to execute apoptosis [75, 76]. In this context, increase of both caspase 9 and DNA fragmentation in asthmatic patients and OVA-induced mice were observed [77]. Further, the disturbance in the balance between apoptotic and antiapoptotic members of Bcl2 has been demonstrated in asthma [78]. It has been shown that exposure of bronchial epithelial cells to TNF-alpha and IFN-gamma leads to the damage of mitochondria and consequently epithelial injury [79]. Supportively, mucous cell apoptosis has been induced by IFN-gamma by BAX upregulation and its translocation into mitochondria [80], indicating that mitochondria-mediated apoptosis could be independent of Th1/Th2 types of inflammation. However, mitochondrial-independent pathways are also likely to be important in epithelial apoptosis. The mitochondrial-apoptotic pathway is also crucial for programmed death of immune cells during resolution of asthma [81, 82]. Interestingly, glucocorticoids have been shown to have proapoptotic effect on human eosinophils via the mitochondrial pathway [83]. Translocation of Bax to the mitochondria, release of cytochrome c, caspase-independent collapse of the mitochondrial membrane, and subsequent activation of caspases are involved in spontaneous eosinophil apoptosis [84]. Failure of inducing eosinophil apoptosis by steroid could be one of the mechanisms of steroid resistance in severe asthmatic patients [85, 86]. This subgroup of asthmatic patients is difficult to treat and consumes a disproportionately large share of health resources [87]. Thus, investigating the role of mitochondria in steroid resistance is an important area of research (Fig. 3.5).
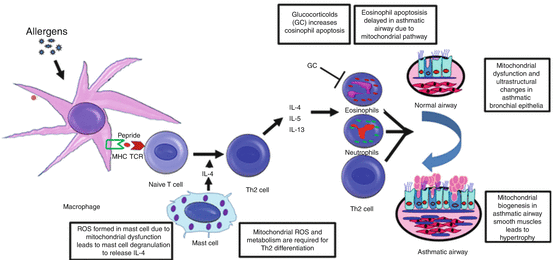
Fig. 3.5
Schematic diagram to show the involvement of mitochondria at various stages of asthma pathophysiology. Mitochondrial dysfunction occurs in mast cells leading to generate ROS which degranulates mast cell to release IL-4 that converts naïve T cells to Th2 cells. These Th2 cells release various cytokines and recruit various inflammatory cells such as eosinophils, neutrophils, and lymphocytes. Recruited inflammatory cells release several proinflammatory mediators which injure airway epithelia. Epithelial injury activates epithelial mesenchymal trophic unit (EMTU) to initiate airway remodeling which consists of hyperplasia and hypertrophy of smooth muscle, goblet cell metaplasia, and subepithelial fibrosis. Asthmatic bronchial epithelia had shown the features of mitochondrial dysfunction, whereas asthmatic airway smooth muscle had shown the features of mitochondrial biogenesis
Airway Remodeling
The important sequelae of bronchial epithelial injury is airway remodeling [88–90]. Repeated allergen exposures lead to structural alterations in the airway, called airway remodeling. Airway epithelia have been postulated to contribute to a large extent to the remodeling process [91]. The airway epithelium in asthmatics is injured by inhaled pollutants, allergens, viruses, and also by endogenous factors such as proteolytic enzymes [89]. Injured epithelium activates epithelial mesenchymal trophic unit (EMTU) [90]. EMTU is active in the lung development at the embryonic stage and it maintains the relationship between epithelia and mesenchymal cells with various growth factors such as TGF-beta, FGF, VEGF, etc. EMTU remains inactive in the adult lung and reactivated by epithelial injury, and its activation in asthmatic airway releases various growth factors which are responsible for various components of airway remodeling. Airway remodeling, a complex phenomenon, consists of alteration in the number and/or type of resident cells and the proportion/quantitation of various components of extracellular matrix [90]. Airway remodeling includes epithelial damage, epithelial hypertrophy and hyperplasia, goblet cell metaplasia, airway myofibroblast transformation, increased airway collagen deposition, hyperplasia, and hypertrophy of airway smooth muscle and bronchial blood vessels [91, 92]. Among these features, hyperplasia and hypertrophy of airway smooth muscle and subepithelial fibrosis are crucial components. On the one hand, there is more of proline causing subepithelial fibrosis [93], and on the other hand, proline leads to inhibit cytochrome c oxidase [94]. Bronchial smooth muscle of asthmatics showed more number of mitochondria with increase in the factors required for mitochondrial biogenesis such as peroxisome proliferator-activated receptor-γ coactivator, nuclear respiratory factor 1, and mitochondrial transcription factor [95].
It was further demonstrated that mitochondria are responsible for smooth muscle proliferation which emphasizes the importance of mitochondria in airway remodeling [95]. Also, mitochondrial biogenesis and increased mitochondrial activity have been reported in asthmatic airway smooth muscle along with altered calcium homeostasis, and it has been suggested that these are crucial in smooth muscle hypertrophy of airway remodeling [95]. However, mitochondrial dysfunction was not observed in those mitochondria from bronchial smooth muscle [95]. In contrast, our lab and others found mitochondrial dysfunction in bronchial epithelia [19, 20]. These indicate the possibility of differential roles of mitochondria in the lung. Thus, it seems there is a differential status of mitochondria in different cell types of asthmatic lungs, and thus careful interpretations are required to understand the precise role of mitochondria in asthma pathogenesis. This is very crucial in converting this knowledge into therapeutics. For example, theophylline causes cAMP-dependent tyrosine phosphorylation of subunit I of cytochrome c oxidase (COX) leading to its inhibition in smooth muscle cells [96]. If this is not cell-specific, it also could inhibit COX in bronchial epithelia and might create havoc in epithelial homeostasis. Thus, meticulous investigations are needed to take mitochondrial research in asthma from bench to the bedside in the form of mitochondrial medicine. It also has been suggested that both mitochondrial dysfunction and mitochondrial biogenesis are interrelated with each other as the latter feature is the possible compensatory mechanism of the former. In addition, it has been suggested that the dense localization of benzodiazepine receptor (BR) on the outer mitochondrial membrane of airway smooth muscle might have a role in asthmatic AHR [97]. Interestingly, airway smooth muscle is known to have high density of mitochondrial benzodiazepine receptor (MBR), a peripheral benzodiazepine receptor (PBR). There are two major types of BR, CBR (central type) and PBR. CBR is present exclusively in neurons, located at the plasma membrane, and regulates chloride channel intrinsic to the GABAA receptor, which is responsible for the antianxiety, anticonvulsant, and muscle relaxant effects of benzodiazepines [97]. PBR is distributed ubiquitously and predominantly located at the outer mitochondrial membrane; due to this reason PBR is generally called as MBR. The exact physiological role of PBR is not clear but has been postulated to relate to mitochondrial heme synthesis and steroid synthesis. Indeed, specific MBR ligands have been shown to affect the airway smooth muscle contraction in basal as well as pharmacologically stimulated conditions [97]. Also, these MBRs are densely localized in various structures in the lung other than airway smooth muscle, such as airway epithelium and submucosal glands. Further studies are needed to better define the potential role of MBR in the modulation of bronchial caliber and in the pathogenesis of asthma.
Calcium Homeostasis and Asthma
It is well known that calcium plays an integrated role in asthma pathogenesis as most of the pathophysiological features such as airway smooth muscle contraction, mast cell degranulation, mucus secretion, recruitment of inflammatory cells, release of proinflammatory mediators from inflammatory cells, and vagal neurotransmission are dependent on calcium signaling. Indeed, calcium channel blockers have been used in asthmatic patients where beta adrenergic receptor agonists were contraindicated or ineffective [98]. Calcium signals also influence most of the functions of mitochondria such as oxidative phosphorylation, apoptosis, etc. [99]. Other than signaling, the mitochondrion acts as an intelligent calcium buffering system since they can sense and fine-tune intracellular calcium levels. Increased intracellular calcium leads to enhanced mitochondrial uptake of calcium via uniporters present in the inner mitochondrial membrane, and this uptake further regulates the calcium release from the plasma membrane and endoplasmic reticulum. Thus, mitochondria can act as buffering organelles in a spatiotemporal manner to maintain the calcium homeostasis inside the cell [100]. This mitochondrial calcium uptake leads to reduction of intracellular calcium, preventing excitotoxicity in cells. However, mitochondrial calcium overload itself may lead to mitochondrial damage and possibly cell death if there is significant damage caused to many mitochondria in a cell [101, 102]. Though calcium signaling is important for both mitochondrial physiology and asthma pathogenesis, detailed studies of this aspect are still lacking. It is to be noted that ORMDL3, one of the very few genes identified through genome-wide association study in asthma, binds and inhibits sarco-endoplasmic reticulum Ca2+ (SERCA) pump and alters endoplasmic reticulum-mediated calcium homeostasis [103].
Viral Infections and Asthma
Respiratory viral infection is one of the risk factors for asthma development in children [104]. Concurrent viral infections in asthma aggravate asthma features, cause acute exacerbations in chronic asthmatic conditions, and also lead to irreversible stage of airway remodeling [105]. Though it is very clear that viral infections caused by respiratory syncytial viruses, and few other viruses of the paramyxoviridae family, worsen airway inflammation and airway hyperresponsiveness, the mechanisms underlying this remain poorly understood. The bronchial epithelium is the major site of viral infections in the airway and participates in innate immune responses to viral infections [106]. Interestingly, mitochondria are suggested as surveillance sites for viruses due to the presence of antiviral proteins in mitochondria [107–109] such as MAVS (named for its mitochondrial antiviral signaling). Nucleic acids of various paramyxoviruses are recognized intracellularly by cytoplasmic receptors such as by RIG-I (retinoic acid-inducible gene I)/melanoma differentiation-associated protein 5 (MDA5) which further binds with MAVS present in outer mitochondrial membrane through CARD-CARD (caspase activation and recruitment domains) interactions [110, 111]. MAVS further recruits other molecules such as Tom70, and TNF-receptor-associated factors to assemble signalosome complex and activate IKK and TBK-1 complexes to signal both NF-κB and IRF3 signaling pathways to further initiate type I interferon responses to evade viral pathogens [110, 111]. In this signaling, mitochondrial localization of MAVS is very crucial as removal of MAVS from mitochondria evades the host immune system. Whether viral infection-induced mitochondrial stress triggers asthma related pathways, or mitochondrial dysfunction in asthmatic airway epithelium accentuates viral infection, remains to be studied.
Therapeutics
The studies discussed above indicate strongly that mitochondrial dysfunction is an important aspect of asthma pathophysiology. It is therefore important to evolve strategies for mitochondrial protection as part of efforts to promote lung health and alleviate asthma. At this time, there are many gaps in the available knowledge, and further investigations are required to develop a sufficient understanding of the interface between mitochondrial biology and lung disease. Numerous efforts are being taken to bring mitochondrial bench research to bedside to control/cure various diseases [112–114]. As a result “mitochondrial medicine” was introduced with the recognition of new target sites in mitochondria. There are two major types of drug targets in mitochondria: direct or primary which acts specifically on mitochondrial enzymes and metabolic pathways and indirect or secondary which acts on mitochondria in nonspecific or accidental manner [115]. Possible reasons for secondary mechanisms are structural homology between mitochondria and other cellular parts, bacterial origin of mitochondria, functional or structural cross talk between mitochondria with other cellular compartments, etc. For example, the secondary effects of various antibiotics like chloramphenicol which affect protein synthesis of both bacteria and mitochondria may be explained by the bacterial origin of mitochondria [116]. The capacity of stem cells in donating mitochondria in rescuing injured airway epithelia in acute lung injury condition indicates another emerging target through the healthy mitochondrial transfer to damaged cells [117]. Though various reports demonstrated the therapeutic capacity of stem cells in asthma [118, 119], whether this is directly through the donation of mitochondria is yet to be demonstrated.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


