Fig. 6.1
ROS-mediated peroxidation of CL forming different molecular species of PUFA hydroperoxides. ROS causes peroxidation of linoleic acid in CL and forms monohydroperoxide and dihydroperoxides on one linoleate ester. Also, in some cases, ROS-induced oxidation of CL leads to the formation of bis-monohydroperoxides on two adjacent linoleate esters
Environmental Metals, Particulate Matter (PM), Lipid Peroxidation, and Lung Mitochondria: Respiratory and Lung Diseases
The airborne trace heavy metals and particulate matter (PM) have been shown to pose a serious threat to the respiratory and lung health of humans through the generation of ROS and oxidative stress [48]. In this context, the role of mitochondria-generated ROS in the pathogenesis of human diseases has been emphasized [49]. Needless to mention, the ROS-induced membrane lipid peroxidation and mitochondrial dysfunction as the underlying mechanisms of tissue damage cannot be ruled out. The heavy metal cadmium (Cd) is an established environmental pollutant, etiological factor in emphysema, and a carcinogen that induces lung cancer [50]. In order to establish the Cd-induced damage of the lung, a study has been conducted which reveals that Cd causes damage of the MRC-5 fetal lung fibroblasts through ROS production, lipid peroxidation, and alterations in the mitochondrial membrane potential [50]. The results of this study confirm that the ROS-mediated lipid peroxidation and mitochondrial alterations are involved in the damage of lung fibroblasts as an underlying mechanism of lung toxicity of Cd. Atmospheric residual oil fly ash (ROFA) PM has been shown to be associated with many environmentally induced lung diseases such as the chronic obstructive pulmonary disease and lung cancer [51]. It has been shown that the ROFA PM causes cytotoxicity to the human alveolar epithelial cells (A549 cells) in vitro through the formation of ROS, DNA damage, lipid peroxidation, and mitochondrial alterations [51, 52]. Thus, the transition metals (vanadium, iron, and nickel) present in the ROFA PM have been identified as potential initiators of mitochondrial dysfunction in the alveolar epithelial cells through oxidative stress (ROS production and lipid peroxidation) that could be critical in the onset and progression of the environmental lung diseases.
The PM air pollution of the urban microcosms is a global health concern due to its association with the respiratory and lung diseases, cardiovascular diseases, morbidity, and mortality among adults as well as children as revealed by the epidemiological studies [53]. Especially, the respirable PM10 has been shown to be associated with the ROS production and oxidative stress, causing inflammation through the activation of critical signaling pathways, and therefore, PM10 is considered an environmental risk factor in the aggravation of respiratory and lung diseases [53]. Airborne PM such as silica, TiO2, nanoparticles, and those generated by the combustion of wood and fossil fuels have been shown to cause the intracellular formation of ROS resulting in oxidative stress, lipid peroxidation, DNA damage, depletion of antioxidants, and alterations of the mitochondrial functions in animal models and humans [54]. The urban airborne PM2.5 collected from the Dunkerque City air in France has been shown to cause cytotoxicity, oxidative stress (lipid peroxidation), DNA damage, mitochondrial dysfunction, and inflammatory response in the human lung epithelial L132 cells in vitro [55]. The PM2.5 has also been shown to cause apoptosis associated with tumor necrosis factor-α secretion, caspase activation, DNA damage, and mitochondrial involvement in the human lung epithelial L132 cells in culture [56]. These studies have revealed that the metals (especially Al, Fe, and Pb) and polycyclic aromatic hydrocarbons (PAHs) present in the urban airborne PM2.5 could be involved in causing oxidative stress (lipid peroxidation) and associated mitochondrial alterations and apoptosis in the lung epithelial cell model [55, 56]. Another study conducted to establish the mechanisms of damage of the lung epithelial cells (A549 cells) induced by the airborne PM2.5 of Abidjan, France, has demonstrated that the PM2.5-induced oxidative damage of A549 cells in culture has been mediated by oxidative stress including the loss of antioxidant defenses, elevated lipid peroxidation, and alteration in the mitochondrial dehydrogenase activity [57]. Also, this study reveals that the Abidajan PM2.5 contains trace metals that are capable of inducing oxidative stress through ROS generation, antioxidant depletion, oxidative stress including lipid peroxidation, and the mitochondrial alterations. Overall these studies suggest the role of ROS and oxidative stress, lipid peroxidation, mitochondrial dysfunction, and apoptosis in the trace metal- and airborne PM-induced cellular damage of the respiratory tract leading to the respiratory and pulmonary diseases including lung cancer [58].
Herbicides and Lipid Peroxidation and Lung Mitochondria: Respiratory and Lung Diseases
Paraquat, the extensively used bipyridium herbicide (1,1′-dimethyl-4,4′-bipyridium dichloride) in addition to damaging several organs, is known to cause oxidative injury to the lung leading to pulmonary fibrosis [59]. Paraquat has been shown to cause O2 − formation through the activation of the mitochondrial NADH-ubiquinone oxidoreductase of the complex I and the production of paraquat radicals, which cause oxidative stress and lipid peroxidation in the lung submitochondrial particles of the paraquat-administered rats [60]. Furthermore, it has been shown that cytotoxicity of paraquat in the rat lung following in vivo administration of the herbicide has been mediated by the mitochondrial dysfunction through complex I activation and lipid peroxidation in the inner membrane [61]. More interestingly, the role of mitochondrial lipid peroxidation in mediating the paraquat-induced lung damage and protection of associated lung fibrosis by the heat-shock protein 60 (HSP60) in the paraquat-intoxicated rats has been observed [59]. Hence, lipid peroxidation of the IMM appears to play a crucial role in lung damage and fibrosis induced by paraquat through oxidant production and oxidative stress.
Carcinogens, Lung Mitochondria, and Lipid Peroxidation: Lung Cancer
Several toxicants including xenobiotic chemicals such as chemical carcinogens are known to cause lung cancer through the mitochondrial lipid peroxidation and oxidative stress [62–64]. The well-established carcinogen benzo[a]pyrene (B[a]P) has been shown to cause elevated extent of the mitochondrial lipid peroxidation along with the lowered antioxidant status and decreased activities of the mitochondrial bioenergetic enzymes in the neoplastic lung tissue of mice [62]. Capsaicin, a natural product occurring in hot peppers (Capsicum annuum) has been shown to protect against the B[a]P-induced lipid peroxidation and alterations in the lung mitochondria and exert chemopreventive action against lung cancer, suggesting that the mitochondrial lipid peroxidation has an apparent role in the chemical carcinogen-induced lung cancer in the mouse model. In another study, the carcinogen B[a]P has been shown to cause the mitochondrial damage in the mouse lung through the ROS formation, lipid peroxidation, loss of antioxidant status, and mitochondrial membrane damage and dysfunction, all of which have been shown to be protected by crocetin, a saffron (Crocus sativus) carotenoid [63]. Since B[a]P is a PAH carcinogen also present in cigarette smoke and known to cause lung cancer, a study has been conducted on the B[a]P-induced lung mitochondrial oxidative damage in vivo in mice and its protection by baicalein, a well-established flavonoid and lipoxygenase inhibitor present in the roots of Scutellaria baicalensis [64]. This study has demonstrated that B[a]P causes the mitochondrial lipid peroxidation and formation of DNA adducts with malondialdehyde (MDA), an end product of lipid peroxidation. Also, this study has revealed that baicalein exhibits chemotherapeutic action in protecting against B[a]P-induced lung cancer possibly through attenuation of the mitochondrial damage caused by lipid peroxidation and formation of MDA-DNA adducts. Another in vivo study in mice has shown that the tobacco carcinogen B[a]P induces lung cancer through oxidative stress, lipid peroxidation, perturbation of antioxidant status, alterations in the electron transport chain activities and energy (ATP) generation, and ultrastructural modifications in the lung mitochondria [65]. Interestingly, hesperidin, a flavanone present in citrus fruits, has been shown to offer protection against the B[a]P-induced lung mitochondrial damage including lipid peroxidation, suggesting the antineoplastic and mitochondrial protective actions of hesperidin in the carcinogen-induced lung cancer. Another in vivo investigation in mice has revealed that elevation of the lung mitochondrial lipid peroxidation and antioxidant depletion appear to play crucial roles in the B[a]P-induced lung cancer [66]. Furthermore, this study has demonstrated the protective actions of piperine (an alkaloid present in the black pepper, Piper nigrum) against the B[a]P-induced lung mitochondrial lipid peroxidation and antioxidant alterations and also against the B[a]P-induced lung cancer. Taken together, these studies highlighted above have demonstrated that the mitochondrial lipid peroxidation and dysfunction are tightly associated with the chemical carcinogen-induced lung cancer, and natural products of plant origin could act as antineoplastic agents in the lung through ameliorating the mitochondrial lipid peroxidation.
Hypoxia, Hyperoxia, Oxidants, and Radiation Modulate Lipid Peroxidation and Lung Mitochondrial Function
Disturbances in lung oxygen concentrations (pO2) such as hypoxia and hyperoxia that generate ROS and exposure to environmental oxidants including nitrogen dioxide (NO2) and ozone (O3) have been shown to cause lung injury wherein the membrane lipid peroxidation and the mitochondrial damage appear as important players [67, 68]. Hyperoxic injury has been established to be mediated by generation of oxygen free radicals, lipid peroxidation, and oxidative stress [69]. Hyperoxic lung injury is not an exception to this mechanism of injury, and antioxidant defense systems are known to protect against the hyperoxia-induced lung damage [70]. GSH has been established as a critical player in maintaining the thiol-redox homeostasis and protection against oxidative injury in the lung [71]. Along those lines, it has been shown that the hyperoxic lung injury in mice in vivo associated with the increased mitochondrial ROS production and lipid peroxidation (8-isoprostane formation) is attenuated by the cellular thiol-redox enhancer/stabilizer, N-acetyl-L-cysteine (NAC), through enhancement of GSH levels and induction of Mn-superoxide dismutase (Mn-SOD) [72]. Thus, in this study NAC has been shown to protect against the hyperoxic lung injury through attenuation of the mitochondrial oxidative stress by enhancing the antioxidant status. Acute hypoxia has also been shown to cause ROS formation and elevation of extent of lipid peroxidation in the lung mitochondria of rats [68]. However, in this study, it has been demonstrated that daily exposure to moderate hypoxia (5 min exposure to 10 % O2) alternatively with hyperoxia (5 min exposure to 30 % O2) for 2 weeks lowers the hypoxia-mediated lipid peroxidation and elevates GSH levels leading to protection of the acute hypoxia-induced mitochondrial damage in the lung. Also, the results of this study suggest that exposure of the lung to alternate cycles of hypoxia and hyperoxia induces adaptive responses for the lung to cope with the oxidant damage during acute hypoxic stress through protection against oxidative stress (lipid peroxidation) in the lung mitochondria.
As discussed earlier, the distinct and intriguing feature of the mitochondrion is its possession of the unique phospholipid CL in the inner mitochondrial membrane which is highly enriched with PUFAs. CL has been unequivocally established as a master switch to initiate apoptosis [73]. Peroxidation of the mitochondrial CL, release of cytochrome c, and induction of apoptosis in several systems have been established and shown to play crucial roles in several human diseases [74–77]. Incidentally, it is rapidly emerging that the oxidant-mediated respiratory and lung diseases are either associated with or mechanistically regulated by the mitochondrial CL peroxidation and associated apoptotic cell death. Utilizing the high-performance liquid chromatography (HPLC) and mass spectrometry (MS) methods, it has been demonstrated that the mitochondrial CL undergoes peroxidation forming hydroperoxy and hydroxy species of PUFA esters and thus contributes to the induction of inflammation and apoptosis in the pulmonary artery endothelial cells and lung tissue under hyperoxic stress and following inhalation of carbon nanotubes [78]. With the use of oxidative lipidomics technology aided by the electrospray ionization MS (ESI-MS), it has been elegantly shown that the hyperoxia-induced acute lung injury in mice in vivo is mediated by peroxidation of the lung mitochondrial CL and non-mitochondrial PS [79]. Furthermore, this study demonstrates that hyperoxia causes peroxidation of CL and PS in the mouse lung endothelial cells leading to apoptosis, which is significantly protected by the mitochondria-targeted free radical scavenger, hemi-gramicidin S conjugate with XJB-5-131(a nitroxide) [79]. Overall, these studies provide convincing evidences supporting the role of mitochondrial CL peroxidation in acute lung injury caused by hyperoxia.
Rats subjected to thoracic γ-ray irradiation have shown elevated lipid peroxidation in the lung mitochondria as the underlying mechanism for radiation-induced interstitial pneumonitis through oxidative stress [80]. This observation has also paralleled with the decreased activities of catalase and SOD in the lung cytosol, suggesting that the radiation-induced mitochondrial lipid peroxidation has been exacerbated by the loss of activities of antioxidant enzymes. The gaseous air pollutant and constituent of cigarette smoke nitric oxide (NO) and the product of its reaction with superoxide (O2 −) peroxynitrite (ONOO−) have been shown to induce oxidative stress and cell death in the lung epithelial cells [81]. Also, this study reveals that the NO-induced lung epithelial cell death is different from that induced by ONOO− as the former and latter being apoptotic and necrotic, respectively. Furthermore, this study suggests that the NO-induced apoptotic cell death of the lung epithelial cells could be due to the mitochondrial damage, cytochrome c release, and caspase activation. The ONOO−-induced necrotic cell death could also be mediated by lipid peroxidation since ONOO− has been observed to induce cellular lipid peroxidation. Vitamin A supplemented at clinical doses as retinol palmitate (1,000–9,000 IU/kg bw/day for 28 days) has been shown to exert prooxidant action in exacerbating the NO-induced lung mitochondrial oxidative and nitrative stress through lipid peroxidation, protein carbonyl formation, and elevated levels of 3-nitrotyrosine formation [82]. Although the nitric oxide synthase (NOS)-specific inhibitor, L-NAME has been observed to offer marginal protection against the vitamin A-induced lung mitochondrial oxidative and nitrative stress, this study suggests that the exact mechanism of exacerbation of the NO-induced lung mitochondrial oxidative and nitrative damage by vitamin A remains elusive.
Tobacco Smoke, Mitochondria, and Lipid Peroxidation
Tobacco smoke including cigarette smoke is known to contain a plethora (>3,800) of toxic compounds that are capable of mediating the free radical-mediated reactions in the respiratory tract leading to lipid peroxidation, oxidative stress, damage of the macromolecules (proteins and DNA), and ultimately lung damage and pulmonary diseases such as emphysema and lung cancer [83, 84]. Therefore, it appears that the mitochondria of lung exposed to tobacco smoke are vulnerable to oxidant-induced damage that is likely to mediate the pathophysiological condition of the organ. The mitochondrial DNA (mtDNA) in humans has been shown to undergo mutations as a result of aging and environmental changes, and the role of oxidative stress in causing mutations of the mtDNA is not ruled out [85]. It has been shown that significant base pair deletion mutations of the mtDNA in lungs of smokers and aging individuals are associated with lipid peroxidation and oxidative damage of DNA [85]. This study underscores the critical role of lipid peroxidation in augmenting oxidative DNA damage in lung tissue of smokers leading to mutations of the mtDNA. It is increasingly becoming evident that environmental factors exacerbate the tobacco smoke-induced respiratory diseases such as asthma through synergistic actions wherein the oxidative stress-mediated lung mitochondrial damage is a critical player [86]. The combined exposure of particulates in printer emissions and environmental tobacco smoke has been shown to cause oxidant (ROS) generation, lipid peroxidation, antioxidant depletion, and mitochondrial damage and dysfunction in the lungs of asthmatic mouse model [86]. Thus, this study has revealed that the environmental factors such as the printer particulate emissions exacerbate pulmonary diseases such as asthma through lipid peroxidation, oxidant damage, and lung mitochondrial damage and dysfunction. Exposure of cigarette smoke extract (CSE) has been shown to cause damage to the human alveolar epithelial cells in vitro [87]. This study has revealed that the CSE-induced alveolar epithelial cell injury (necrosis and apoptosis) are associated with lipid peroxidation and altered mitochondrial membrane potential suggesting that the lipoperoxidative mitochondrial damage and dysfunction in the alveolar epithelial cells appear to play important roles in the cigarette smoke-induced lung damage and diseases such as emphysema.
Lipid Peroxidation Products, Eicosanoids, and Lung Mitochondria
PUFAs of membrane lipids undergo free radical-mediated peroxidation in the living cells leading to the formation of diverse and highly reactive compounds including the fatty acid hydroperoxides, aldehydes, and electrophilic carbonyls which not only react with cellular macromolecules (proteins and nucleic acids) but also cause cellular dysfunction and damage. The mitochondria have been highlighted as both originators and targets of toxic products of lipid peroxidation in heart, especially under pathophysiological conditions [88]. Hence, it is surmised that the reactive products of lipid peroxidation can also cause the mitochondrial dysfunction and damage in the respiratory tract including lung thus contributing to the respiratory and lung diseases (Fig. 6.2). Linoleate hydroperoxide (LOOH) has been shown to inhibit the agonist-induced O2 − formation and cause the mitochondrial membrane alterations in the rat alveolar macrophages [89]. Hence, it is possible that PUFA hydroperoxides such as the LOOH commonly formed in the alveolar macrophages by either enzymatic or nonenzymatic oxidation of linoleate of the cellular membranes may be involved in the macrophage-mediated lung disorders and diseases. Pulmonary surfactant has been shown to attenuate the LOOH-induced lipid peroxidation in the isolated rat lung mitochondria suggesting the role of lipoperoxidative damage of the lung mitochondria and its protection by the surfactant proteins associated with acute respiratory distress syndrome (ARDS) [90]. The air pollutant and cigarette smoke constituent acrolein (a strong electrophilic and toxic α,β-unsaturated aldehyde) has also been shown to be formed from PUFA peroxidation in the cells that is involved in the pathogenesis of several diseases including the respiratory diseases such as asthma, chronic obstructive pulmonary disease (COPD), lung cancer, and cystic fibrosis [91–93]. In the human small airway epithelial cells (HSAEpCs), acrolein has been demonstrated to induce cytotoxicity and apoptosis through ROS generation, oxidative stress, apoptotic signaling pathway activation, and cytochrome c release from the mitochondria [93]. The acrolein-induced damage of HSAEpCs has been shown to be protected by inhibiting aldose reductase, an enzyme that is involved in glucose metabolism possibly through suppressing the reactivity of acrolein with GSH attenuating the formation of acrolein-GSH adducts. From this study it is clear that the PUFA lipid peroxidation product acrolein causes damage to the airway epithelial cell mitochondria leading to the apoptotic cell death. This is also supported by another study which has demonstrated that acrolein causes cytotoxicity and the mitochondria-mediated apoptosis in human lung cancer A549 cells [94].
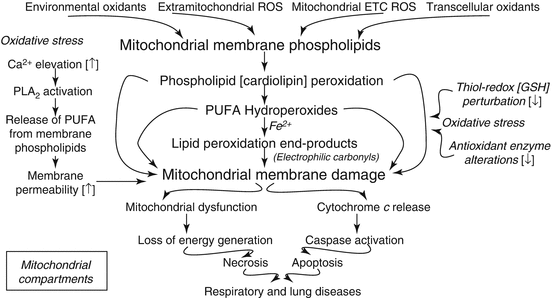
Fig. 6.2
Role of eicosanoids in causing mitochondrial damage and apoptosis in lung cells that are involved in respiratory and lung diseases
The role of oxLDL has been recognized in the pathogenesis of atherosclerosis and associated cardiovascular diseases. However, oxLDL has also been identified as an inducer of the mitochondrial ROS generation in the vascular endothelial cells (ECs), and the underlying mechanism is yet to be established [95]. Thus, the metabolically formed and highly reactive peroxidized oxLDL is capable of inducing ROS generation in the mitochondria that has been shown to be associated with the vascular EC dysfunction, activation, and inflammation in conjunction with the pathogenesis of atherosclerosis. Although it is farfetched, it is probable that oxLDL could be involved in the pathogenesis of certain respiratory and lung diseases through the generation of ROS in the mitochondria of lung vasculature which warrants further in depth studies.
Lipid oxygenases such as cyclooxygenases (COXs) and lipoxygenases (LOXs) are known to oxidize PUFAs, especially arachidonic acid released from the membrane PLs upon the action of phospholipase A2 leading to the formation of bioactive eicosanoids including the prostaglandins (PGs), PUFA hydroperoxides (PUFA-OOH), and leukotrienes (LTs). These eicosanoids exert diverse physiological actions in the cells leading to both cell injury and cytoprotective responses. However, it is beginning to emerge that the eicosanoids also exhibit profound mitochondria-affecting actions in the lung. In the A549 lung cancer cell line, it has been shown that the PGs apparently induce apoptosis through the regulation of expression of the PGE2 receptors (EP receptors) and PGF2α receptors (FP receptors), especially the former being the only one observed in the mitochondria and both types of receptors being present in the plasma membrane [96]. This study demonstrates that the COX-generated PGs are involved in the lung cancer cell apoptosis through regulation of expression of the EP and FP receptors in the cell and mitochondrial membranes. COX-2, an isoform of the COX family of enzymes, is known to regulate the tumor growth, and therefore a study has been conducted to demonstrate the localization of COX-2 in several types of cancer cells including the lung cancer A549 cell line [97]. This study reveals that the presence and co-localization of COX-2 and heat-shock protein 60 (HSP60) in the mitochondria of A549 lung cancer cells. Furthermore, this study suggests that COX-2 may be involved in the cancer cell resistance to apoptotic death. The cyclopentenone class PGs (PGJ2s), especially the 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), originating from dehydration of the COX-generated PGD2, are highly bioactive and mediate important cellular functions including progression of cell cycle, expression of HSP, cell growth and differentiation, anti-inflammatory response, cytoskeletal alterations, changes in redox status, protein synthesis, and apoptosis [98]. More importantly, the elevated levels of 15d-PGJ2 formed from the COX-catalyzed arachidonic acid metabolism during several disease conditions including inflammation has been recognized [98]. The cyclopentenone PGJ2s including 15d-PGJ2 contain the α,β-unsaturated carbonyl group and are electrophilic in nature, and therefore they form covalent adducts (Michael addition) with nucleophiles (-SH group) including cysteine and GSH in a non-receptor mechanism leading to the regulation of redox-dependent cellular signal transduction cascades [98]. In the human non-small cell lung carcinoma cells (A549 cells), 15d-PGJ2 has been observed to induce apoptosis through the mitochondrial cytochrome c release [99]. Thus, this study reveals that the cyclopentenone PG induces apoptotic cell death in the human lung A549 cancer cells through activation of the mitochondrial pathway of apoptosis.
The LOX-generated eicosanoids have been shown to be bioactive and elevated during the inflammatory respiratory diseases such as asthma [100]. In the human lung A549 cells, interleukin-4 (IL-4) has been shown to cause the upregulation of 15-LOX that catalyzes the formation of 15(S)-hydroxyeicosatetraenoic acid (15(S)-HETE), which induces apoptosis in the cells upon binding to the peroxisome proliferator-activated receptor γ (PPARγ). This study also reveals the involvement of mitochondria (cytochrome c release, translocation of the cytoplasmic Bax protein to mitochondria) in the IL-4-induced apoptotic death of the A549 cells through the actions of 15(S)-HETE which may have implications in the pathogenesis of chronic asthma [100]. Exposure of mice and cultured alveolar macrophages to inhalable carbonaceous airborne particles with high electron spin density (free radical characteristics) have shown inflammation, oxidative stress, and mitochondrial abnormalities in the alveolar macrophages in vivo, which are associated with enhanced NO production in lavage fluid of the lung and elevated nitrotyrosine formation in the lung tissue [101]. More interestingly, this study reveals that the alveolar macrophages in culture upon exposure to the carbonaceous airborne particles with high electron spin density exhibit elevated levels of secreted LTB4, an eicosanoid formed from the LOX-catalyzed metabolism of arachidonic acid, suggesting the association of airborne particle-induced mitochondrial damage and LTB4 secretion in the alveolar macrophages. Also, the role of LOX-, COX-, and cytochrome P 450-generated eicosanoids in induction of the mitochondria-mediated apoptosis is emphasized [102]. Taken together, the above highlighted studies have clearly demonstrated that the PUFA peroxidation products generated by nonenzymatic and enzymatic catalysis are bioactive in causing the mitochondrial dysfunction that leads to the cellular and tissue damage of the lungs with implications in the pathogenesis of respiratory diseases (Figs. 6.2 and 6.3).
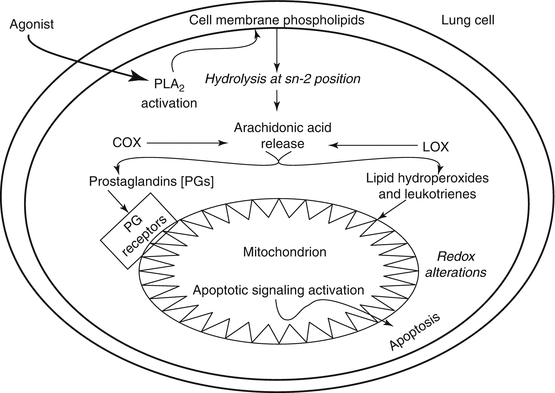
Fig. 6.3
Mechanism of ROS-induced membrane lipid peroxidation leading to mitochondrial dysfunction, necrosis, and apoptosis in lung cells as underlying mechanism(s) of respiratory and lung diseases
Mitochondria-Targeted Free Radical Scavengers and Antioxidants and Protection Against Lipid Peroxidation
The search for effective free radical scavengers and antioxidants to inhibit oxidative stress and lipid peroxidation is a continuous and evolving endeavor in the arena of oxidant biology [11]. The use of water-soluble compounds (e.g., vitamin C and thiol protectants such as NAC) and lipid soluble antioxidants (vitamin E and phytochemical polyphenols and natural products) has been in practice to intervene oxidative stress and protect against oxidant injury in biological systems including the oxidative lung damage [103–106]. However, targeting specific cellular sites such as mitochondria which are both sources and targets of ROS and oxidative stress with effective free radical scavengers and antioxidants is a challenging task. In this regard, the cell-permeable Szeto-Schiller (SS) peptides have emerged as the novel mitochondria-targeted peptide antioxidants which could offer promise in protecting against the oxidant-induced mitochondrial lipid peroxidation and damage [107–109]. In order to effectively target the specific mitochondrial membranes and compartments for effective antioxidant action and protection against the ROS production and oxidant-induced CL peroxidation in mitochondria and apoptosis, strategies such as designing and synthesizing nitroxide-natural product conjugates and nitroxide-gramicidin conjugates have been ingeniously proposed [110]. Especially, the delivery and targeting mitochondrial locations (compartments) precisely with antioxidants (small molecules and enzymes) with the use of efficient triphenylphosphonium and hemi-gramicidin S cargoes have been emphasized [111]. However, dual action compounds that are equally effective as antioxidants (scavenging free radicals and inhibiting lipid peroxidation) specifically in the mitochondria and as apoptosis inhibitors are an unmet need for the therapeutic intervention of oxidant lung damage and oxidative stress-mediated respiratory and lung diseases wherein the mitochondria are at the epicenter.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


