Fig. 8.1
Mitochondria as oxygen sensor. Mitochondrial reactive oxygen species generation at complex III of the respiratory chain. IMS intermembrane space, SOD superoxide dismutase
Mitochondria in Cancer
One constitutive metabolic characteristic of cancer cells is the upregulation of glucose metabolism and the dependence on anaerobic glycolysis for the synthesis of ATP, even in the presence of abundant oxygen. This phenomenon was first described by Warburg in 1930 [36], and it is accompanied by a durable shutdown of mitochondrial respiration that allows cancer cells to thrive in the hypoxic environments characteristic of solid tumors as the lesion grows progressively further from the blood supply [37, 38]. Moreover, this switch to aerobic glycolysis in cancer, the so-called Warburg effect, is the basis of positron emission tomography in which a glucose analog tracer (2-18fluoro-2-deoxy-D-glucose) differentiates between normal and tumor tissue by their avidity for glucose [36] (Fig. 8.2).
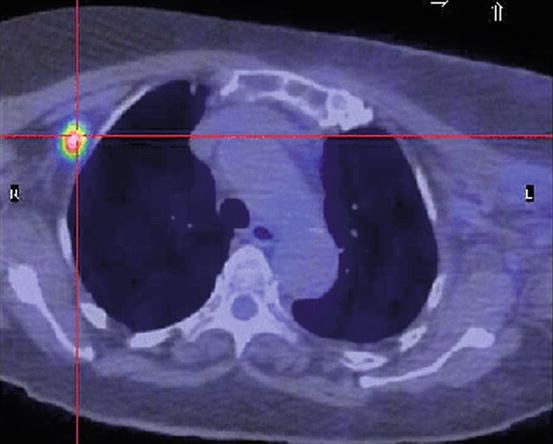
Fig. 8.2
Positron emission tomography. The switch to aerobic glycolysis in cancer, the so-called Warburg effect, is the basis of positron emission tomography in which a glucose analog tracer (2-18fluoro-2-deoxy-D-glucose) differentiates between normal and tumor tissue by their avidity for glucose
The mechanisms for the abnormal regulation of energy metabolism in cancer cells seem to be mediated by mitochondrial DNA mutations causing the impairment of the respiratory chain functioning [39, 40]. Some of these mutations inhibit oxidative phosphorylation and enhance the production of ROS and thus promote tumor cell proliferation [39]. Another category of milder mutations could permit tumors to adapt to new microenvironments, especially when tumors progress and metastasize [39]. In most cases such mutations are acquired during or after oncogenesis; however, certain mitochondrial DNA mutations have been shown to provide a genetic predisposition to cancer development [39]. The known mechanisms by which these mutations lead to oncogenicity are dual [36]. In the first place the inactivation of the mitochondrial metabolic pathways endangers anaerobic glycolysis, but also certain mutations lead to the stabilization of HIF-1α and generate a pseudohypoxic state accompanied by HIF-dependent reprogramming of the metabolism toward aerobic glycolysis providing a gain of function in terms of the activation of hypoxia responses [37, 41]. The paradigm is in the case of some mutations affecting subunit B, C, or D of succinate dehydrogenase (SDH) causing pheochromocytoma and in the case of fumarate dehydrogenase leading to leiomyoma, leiomyosarcoma, or renal carcinoma [36]. The loss of activity of these enzymes results in the accumulation of succinate or fumarate in the cytosol, respectively. This, in turn, favors the activation of HIF [37]. An alternative mechanism has been proposed for the mutations of SHD subunits. It suggests that when any other subunits of SDH are inactivated, the A subunit SDH causes an increase in ROS signaling, which stabilizes HIF-1α [41] by the mechanisms described above (see hypoxic pulmonary vasoconstriction). Some of these mutations of mitochondrial DNA might be useful tumor markers, with the advantage that are easier to determine since in the cell there are higher number of copies of the mitochondrial genes [42].
Another feature of mitochondria from cancer cells is their resistance against the induction of mitochondrial permeability transition (MPT), a process that mediates the intrinsic pathway of apoptosis [43–45]. In cancer cells MPT is regulated by proteins from the Bcl-2 family [46], the proteins forming the permeability transition pore (PTP) complex [47], proteins that affect mitochondrial dynamics (fusion and fission) [48, 49], and transcription factors (such as the tumor suppressor protein p53) [50]. MPT inhibition resulting in disabled apoptosis is important for the development of tumors [51]. Interestingly progression and resistance to therapy due to continued smoking may be due to the effects of nicotine on its ability to inhibit chemotherapy-induced apoptosis in lung cancer cells [45]. Thus, the mitochondrial alterations have clinical interest and are being the target of novel anticancer therapies [9, 36, 52] illustrating how the slow accumulation of fundamental knowledge eventually generates medically exploitable information.
Epithelial Cell Apoptosis and Lung Remodeling
Apoptosis may play an important role in lung diseases in two different ways. First, failure to clear unwanted cells by apoptosis will prolong the inflammation because of the release of their toxic contents and also delay repair processes [53]. Apoptotic cells should be quickly recognized and ingested by phagocytes before releasing their toxic contents, unlike accidental cell death or necrosis. Second, excessive apoptosis may cause diseases [54] (Fig. 8.3). There are two major signaling pathways of apoptosis. The extrinsic or receptor-mediated apoptosis is triggered by the binding of certain ligands such as FasL or the tumor necrosis factor (TNF) to cell death receptors on the surface [53, 55, 56]. Most cells in the human body have two receptors for TNF: TNF-R1 and TNF-R2. The binding of TNF to TNF-R1 has been shown to initiate the pathway that leads to caspase activation via the intermediate membrane proteins TNF receptor-associated death domain (TRADD) and Fas-associated death domain protein (FADD) [56]. The other receptor is Fas (CD95). When its ligand, FasL, binds to Fas, the intracellular death domain of Fas is in its turn combined with FADD. Once assembled, FADD leads to caspase-8 activation, which in its turn triggers the caspase cascade [43, 55]. The intrinsic pathway is triggered by a variety of stimuli (i.e., drugs, radiation, infectious agents and ROS, nitric oxide, high levels of calcium, as those induced by prolonged cell hypoxia, or the conformation shift of the cytosolic protein Bax) and involves the formation and opening in a large conductance of the mitochondrial PTP, a nonspecific multiconductance channel of the outer membrane [43] (Fig. 8.4). After cytochrome c is released into the cytosol from the mitochondria, it binds to Apaf1 and ATP, which then activate caspase-9 [43, 55]. In this section we will deal with the information on intrinsic (i.e., mediated by mitochondria) apoptosis in lung disease.
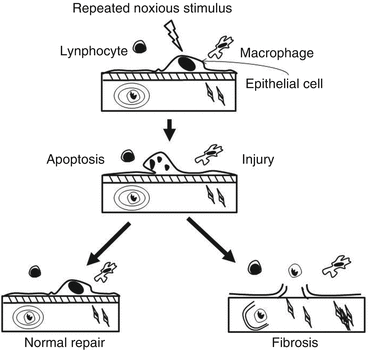
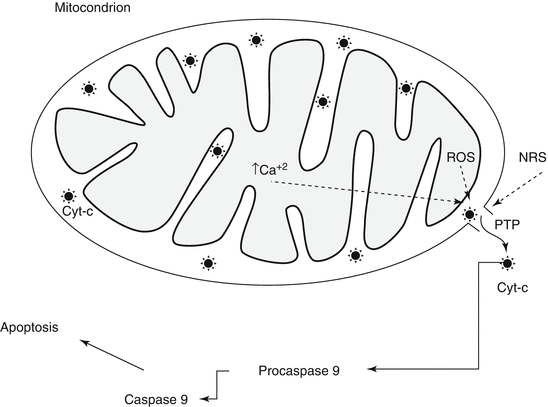
Fig. 8.3
Fibrotic mechanism. Depending on the degree of lung injury, the damaged tissue is normally repaired or to irrepairable damage and remodeling
Fig. 8.4
Intrinsic mechanism of apoptosis. The intrinsic mechanism of apoptosis starts by the formation of the permeability transition pore (PTP) in response to several stimuli (ROS, NRS, high calcium levels, and radiation). The high-conductance pore allows the release of cytochrome c and other facts that initiate the caspase cascade by activating procaspase-9 into caspase-9. ROS reactive oxygen species, NRS nitrosative reactive species, Cyt-c cytochrome c
Acute Lung Injury
Apoptosis of type I alveolar epithelial cells and endothelial cells are observed in acute respiratory distress syndrome (ARDS) [57], and the number of epithelial cell apoptosis is associated with the prognosis of patients with diffuse alveolar damage [58, 59]. The expression of Bax protein is upregulated in alveolar epithelial cells in acute lung injury [58, 59], and lipopolysaccharide, an important factor in acute lung injury, induces apoptosis of endothelial cells of systemic organs in mice [60]. Cytokines with antiapoptotic properties such as IL-6, IL-11, IL-15, and granulocyte-macrophage colony-stimulating factor attenuate hyperoxic lung injury, and this protection is associated with a marked decrease of apoptosis [61–64]. These findings suggest that apoptosis plays a major role in the development of acute lung injury in mice and ARDS in human.
Interstitial Lung Diseases
Alveolar epithelial damage is an important initial event in idiopathic pulmonary fibrosis (IPF) which includes an augmented epithelial cell apoptosis [65–67]. Both the Fas/FasL- and mitochondria-mediated apoptotic pathways are activated in IPF [68]; however, some evidence suggests that Fas-mediated pathway is more relevant [69].
Bronchial Asthma
The loss of columnar epithelial cells is a characteristic feature of asthma. Asthmatic bronchial epithelium is susceptible to H2O2-induced apoptosis [70]. Such susceptibility may contribute to the rising trends in asthma associated with air pollution and diets low in antioxidants [70]. In severe asthma, there is a greater level of apoptotic activity and increased cellular proliferation in the airway epithelium [71]. Since the remodeling processes of bronchial epithelium are associated with various molecules which are involved in epithelial cell apoptosis, treatments targeting epithelial cell apoptosis may be effective in these patients.
Chronic Obstructive Pulmonary Disease
Increased numbers of apoptotic alveolar, bronchiolar, and endothelial cells are frequently observed in lung tissues from patients with COPD [53]. Apoptosis has been associated with the initial injury and defective repair in COPD and the destruction and loss of alveolar structures characteristic of pulmonary emphysema [72, 73]. Since apoptosis is likely to be involved in not only the destructive phase but also remodeling process, regulation of apoptosis may become an effective treatment against COPD.
Cigarette Smoke
Cigarette smoke induces epithelial cell apoptosis [74]. Moreover, it impairs the removal of apoptotic cells [75], and the failure to remove is thought to be also important as a contributor to COPD [76]. CS also causes mitochondrial dysfunction by blocking mitochondrial respiratory chain and decreasing ATP generation which leads to cellular death (apparently necrosis rather than apoptosis) [77]. The heme oxygenase-1 (HO-1) mRNA expression was elevated in the lungs of mice chronically exposed to cigarette smoke. The mitochondrial localization of HO-1 in bronchiolar epithelial cells was confirmed using electron microscopy. Overexpression of HO-1 inhibited cigarette smoke extract induced [78].
Skeletal Muscle Dysfunction of COPD
There is compelling evidence showing the involvement of mitochondria in the muscle dysfunction (SMD) associated to COPD. Typically the citric acid cycle (Krebs cycle) enzyme activity (such as citrate synthase, SDH) and β-hydroxyacyl-CoA dehydrogenase (an enzyme involved in β-oxidation of fatty acids) are lower in patients with COPD as compared with control individuals [79–82]. Conversely, cytoplasmic glycolytic enzyme activities are elevated [79–82]. Patients with mild or moderate COPD usually show a reduced “vastus lateralis” state 3 mitochondrial oxygen consumption, acceptor control ratio, and ATP production [83–86]; however, when normalized by citrate synthase (CS) activity, a measure of mitochondrial density, differences between COPD and normal individuals in mitochondrial oxygen consumption, and ATP production have been found small [84] or nil [85]. In keeping with these findings, measurement of mitochondrial density has been found lower in COPD than in age-matched, healthy controls [87, 88]. The reduction of mitochondrial density and oxidative fiber loss is the typical physiological response of muscle [89, 90]. Moreover, supporting the role of deconditioning, peripheral muscle oxidative capacity has been shown to improve with training [81, 91, 92] although not at the same levels observed in healthy subjects [93].
Conversely to what occurs in the lower limb muscles, the activities of the oxidative enzymes such as CS, SDH, and δ-hydroxyacyl-CoA dehydrogenase are elevated in the diaphragm of patients with severe COPD [94–96], whereas the activities of the glycolytic enzymes are decreased [97]. Enhanced mitochondrial state 3 respiration has also been reported in the diaphragm of very severe COPD patients, and significant correlations were found between the maximal oxidative capacity and patients’ pulmonary indexes of obstruction [98]. These data indicate that the changes in oxidative capacity and mitochondrial function in the diaphragm are in the opposite direction than in the “vastus lateralis” and they appear to become more marked with the progression of the obstruction. This likely reflects a training effect of the muscles [94, 95, 97].
Excessive ROS production is another feature of SMD in COPD. The rates of generation of ROS and other free radicals are elevated in patients with COPD during exercise [99, 100], and mitochondria have been shown to be major contributors [30, 84]. The sources of these oxygen free radicals are complexes I and III, but only those ROS generated by complex III appear to be able to reach the cytoplasm [30, 101]. The oxidative stress associated to excessive ROS production affects membrane lipids, proteins inside and outside the mitochondria, and mitochondrial DNA [88, 101–103] impairing mitochondrial oxidative function by jeopardizing the integrity of the electron transport chain and uncoupling oxidative phosphorylation by decreasing the transmembrane potential [43, 104, 105]. Protein oxidation may modify the structure or chemical properties of the proteins affected [102, 106] and cause a decline in protein function or even complete protein unfolding in muscle cells [101, 104]. Among the proteins affected are the myofibrillar muscle protein that becomes more susceptible to proteases [107], thus contributing to the dysfunction of the muscle.
Conclusions
The mitochondrion of the eukaryotic cells is not only the powerhouse of the cell that generates energy but is also involved in several important cellular functions including cell signaling and death. The mitochondrion also acts as the oxygen sensor especially during hypoxia and signals the information toward the generation of ROS that act as mediators of cell signaling. This hypoxia-induced ROS signaling during hypoxia is believed to be responsible for hypoxic vasoconstriction, activation of HIF, and internalization of the sodium-potassium pump of the alveolar epithelium. In cancer cells, mitochondrial DNA mutations lead to compromised oxidative metabolism and decreased susceptibility to apoptosis that are crucial for survival of the cancer cell. One of the two major signaling pathways of apoptosis involves the mitochondria. Apoptosis is an important player in several lung diseases that contributes to the failure of clearance of unwanted cells and prolonged inflammation and thus is involved in the pathogenesis of the lung diseases. The skeletal muscle dysfunction associated with COPD involves the loss of oxidative capacity, mainly due to the loss of mitochondrial density.
References
2.
McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16:R551–60.PubMed
4.
Gusarova GA, Dada LA, Kelly AM, et al. Alpha1-AMP-activated protein kinase regulates hypoxia-induced Na, K-ATPase endocytosis via direct phosphorylation of protein kinase C zeta. Mol Cell Biol. 2009;29:3455–64.PubMedCentralPubMed
5.
Comellas AP, Dada LA, Lecuona E, et al. Hypoxia-mediated degradation of Na, K-ATPase via mitochondrial reactive oxygen species and the ubiquitin-conjugating system. Circ Res. 2006;98:1314–22.PubMed
6.
Vadasz I, Dada LA, Briva A, et al. AMP-activated protein kinase regulates CO2-induced alveolar epithelial dysfunction in rats and human cells by promoting Na, K-ATPase endocytosis. J Clin Invest. 2008;118:752–62.PubMedCentralPubMed
8.
Skuli N, Liu L, Runge A, et al. Endothelial deletion of hypoxia-inducible factor-2alpha (HIF-2alpha) alters vascular function and tumor angiogenesis. Blood. 2009;114:469–77.PubMedCentralPubMed
9.
Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95:7987–92.PubMedCentralPubMed
10.
Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–65.PubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


