A genetic basis underlies a majority of nonischemic cardiomyopathies (Chapters ). In most cases of cardiomyopathy in adults, there are multiple genes and mutations that are associated with a particular phenotype (i.e., dilated, hypertrophic, arrhythmogenic, or noncompaction). In many examples, however, specific mutations or genes are associated with cardiomyopathy that typically presents at an early age and has extracardiac manifestations.
Although there is an overlap between familial cardiomyopathy and those associated with specific syndromes, the current chapter focuses on diseases that have a genetic basis, that often have extracardiac manifestations, that often occur in infants and children, and that are known by the specific mutation or by a specific eponym. The practice of renaming cardiomyopathy after a new mutational finding (e.g., desminopathy, laminopathy) has been discouraged.
1 Cardiomyopathies that are not defined by the mutational status or clinical syndrome but rather by cardiac clinicopathologic features are discussed in previous chapters.
Noonan Syndrome
Noonan syndrome is an autosomal dominant condition characterized by short stature, heart defects, pectus excavatum, webbed neck, learning problems, cryptorchidism, and facial dysmorphism.
2 It occurs in up to 1 in 1,000 live births.
3 Cardiovascular manifestations occur in 20% of patients and include cardiac hypertrophy (hypertrophic cardiomyopathy [HCM]-like phenotype), valvular dysplasias, atrial septal defect, ventricular septal defect, and patent ductus arteriosus (see
Chapter 3).
Noonan syndrome and related conditions are caused by the dysregulation of the RAS-MAPK signaling pathway. Overall, mutations are found only in a subset of patients and include variants in
PTPN11 (about 50% of identified mutations),
SOS1 (13%),
RAF1 (5% to 15%), and
RIT1 (9%). Mutations of
KRAS,
NRAS,
SHOC2,
BRAF,
MEK1, and
CBL have also been reported.
2,3Pulmonary valve stenosis is the most common cardiovascular manifestation. The rate of HCM-like phenotype is as high as 70% in mutation-positive individuals and is frequently associated with pulmonary valve dysplasia with stenosis. There may be associated atrial and ventricular septal defects and mitral valve disease in up to one-third of these patients.
3The pathologic cardiac manifestations include generalized left ventricular hypertrophy and dome-shaped dysplastic pulmonic valves. Autopsy studies are few, and the pathologic features of the cardiomyopathy have not been studied.
4Myofiber disarray can occur in association with fibrosis (
Fig. 160.1), but classic whorled appearance of sarcomeric HCM is not generally
seen. Magnetic resonance imaging has demonstrated a subset of patients with myocardial bridges and left ventricular diverticula, crypts, and crevices.
5
Friedreich Ataxia
Friedreich ataxia is the most common cause of inherited childhoodonset ataxia. It is an autosomal recessive disease caused by mutations in the human frataxin gene,
FXN. Frataxin functions as a mitochondrial iron chaperone protein. Mutations result in abnormal accumulation of intramitochondrial iron, mitochondrial dysfunction, and oxidative damage.
6,7Cardiac manifestations of Friedreich ataxia occur at an older age than do the neurologic symptoms, generally in early adulthood. Up to 90% of patients will demonstrate cardiac abnormalities on echocardiography. Approximately 20% of patients will develop a hypokinetic dilated cardiomyopathy phenotype that may be fatal. Earlier phases of the cardiomyopathy are characterized by cardiac hypertrophy, which may be asymmetric or concentric.
The severity of left ventricular hypertrophy is related to the number of GAA repeats in the first intron of
FXN. Pathologically, the heart is dilated and hypertrophied.
6 Histologically, there is diffuse interstitial fibrosis, patchy chronic inflammation, and endocardial fibrosis.
6 Small-vessel disease of intramural coronary arteries has also been described.
8Cardiomyopathy in patients with Friedreich ataxia may be exacerbated by diabetic ketoacidosis and Graves disease.
9 Sudden death has been reported as the initial manifestation of cardiac symptoms with diagnosis made by postmortem molecular testing.
6
Myotonic Dystrophy
Myotonic dystrophy is an autosomal recessive disease involving facial, neck, and distal limb muscles. There are two types with differing genetic etiologies. Type 1 (Steinert disease) is caused by mutations in DMPK. Type 2 (proximal myotonic myopathy) is a milder form of the disease caused by mutations in ZNF9.
Cardiac involvement manifests most commonly as conduction defects, which may occur in the absence of severe skeletal disease and occur in the majority of patients. First-degree heart block is the most common abnormality, followed by ventricular tachycardia. Rarely, the progression of the disease can lead to sudden, unpredictable death. Conduction electrophysiologic studies are generally performed to guide implantation of defibrillators.
10Histologic findings are nonspecific. There is typically fibrosis of the bundle of His and proximal bundle branches.
11 Myocytes show nonspecific hypertrophy with occasional vacuoles and disarray.
11 Electron microscopic findings include prominent I bands and myofibrillar degeneration.
12
X-Lined Muscular Dystrophy
Duchenne and the milder Becker muscular dystrophies are caused by mutations in the dystrophin gene DMD, located at Xp21. Dystrophin is a cytoskeletal protein that provides structural support to the myocyte and links the sarcomeric contractile apparatus to the sarcolemma and extracellular matrix.
Duchenne muscular dystrophy (DMD) is most commonly caused by deletion of one or more exons, with duplications, single nucleotide variant, and splicing variants causing a lower percentage of total cases. In patients that survive past the age of 18, development of cardiomyopathy (dilated phenotype) is the rule. In fact, with improvements in treatment for respiratory and other complications, the cardiomyopathy is thought to account for most deaths in DMD (either heart failure or arrhythmia). Pathologically, there is myocardial fibrosis that is typically interstitial, although transmural scars may occur. The changes are most marked in the posterobasal region and adjacent lateral wall of the left ventricle.
13In ˜6% of men with idiopathic dilated cardiomyopathy without skeletal myopathy, defects in the dystrophin gene may be identified.
14 Overall, X-linked inheritance accounts for between 2% and 5% of familial cases of dilated cardiomyopathy that manifests in young men with rapidly progressive heart failure.
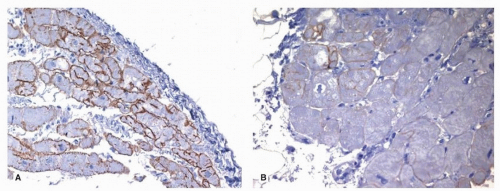
15The presence of increased serum creatine phosphokinase (SCPK) is suggestive of the disease in men with a history of X-linked cardiomyopathy. Mutational analysis for pathogenic mutations in the dystrophic gene DMD is diagnostic, and immunohistochemical staining for dystrophin may also be helpful (
Fig. 160.2).
Female carriers develop a milder form of cardiomyopathy, usually in their 50s. Immunohistochemical stains for dystrophin reveal a patchy loss of dystrophin, with scattered myocytes showing no staining in a mosaic pattern.
16 Cardiac findings are similar to those in males, with biventricular dilation and diffuse fibrous replacement. 16 The right ventricular free wall may become thin and replaced by fibrofatty tissue, mimicking arrhythmogenic right ventricular cardiomyopathy.
16Becker dystrophy is also the result of mutations in the
DMD gene, but unlike DMD, some protein is produced. These individuals usually manifest a milder phenotype, but most (70%) will also develop cardiomyopathy, which, like DMD, is a common cause of mortality.
17
Cardiac Involvement in Emery-Dreifuss and Myofibrillar Myopathies/Muscular Dystrophies
Emery-Dreifuss muscular dystrophy is a myopathy that causes cardiac dysrhythmias, late-onset heart failure, and progressive skeletal myopathy characterized by contractures of the neck, elbows, and ankles. It was generally considered an X-linked disease, although autosomal dominant forms have been more recently described. It is an example of a nuclear “envelopathy,” or “laminopathy” caused by mutations of genes that encode proteins of the nuclear envelope. Mutations in the
EMD gene, encoding emerin, cause the X-linked form, while
LMNA gene mutations are responsible for autosomal forms, which manifest as a dilated phenotype (see
Chapter 19).
18Mutations in
LMNA also cause a related condition, limb girdle myotonic dystrophy type 1B.
19 Endomyocardial biopsy specimens show only nonspecific findings.
20The “myofibrillar myopathies” (see
Chapter 27) are characterized by myofibrillar disorganization of the Z-disk progressing to myofibrillar degradation in the adjacent sarcomere.
21 The pathologic diagnosis of myofibrillar myopathy is established by skeletal muscle biopsy in which hyaline deposits are present that may be congophilic. Cardiac involvement is most frequent in myofibrillar myopathies caused by mutations in desmin and zaspin (so-called desminopathy and zaspopathy, respectively).
Barth Syndrome
X-linked dilated cardiomyopathy in male infants is most frequently caused by Barth syndrome (dilated cardiomyopathy, skeletal myopathy, growth retardation, and neutropenia).
22 Male neonates or young infants present with congestive heart failure, neutropenia, and 3-methylglutaconic aciduria. Children die in infancy from progressive heart failure, sudden death, or sepsis. The pathologic findings are nonspecific, but imaging studies demonstrate that at least one-half of patients have left ventricular noncompaction.
23Barth syndrome is caused by mutations in the tafazzin gene (
TAZ) on chromosome Xq28. Heart failure is common as patients survive childhood and is caused by cardiomyopathy with both a dilated and noncompaction phenotype, as well as endocardial fibroelastosis (EFE).
24 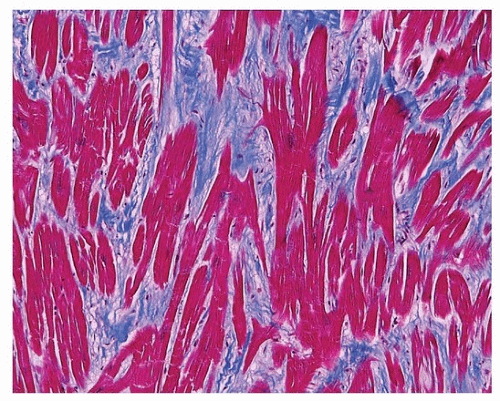


 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access
