Fig. 62.1
miRNAs are a distinct class of regulatory molecules. MicroRNAs are transcribed from the genome as pri-miRNAs (1) that are processed in the nucleus by a protein complex (2) containing the DGCR8 and the RNase Drosha as key molecules. The resulting hairpin-structured pre-miRNAs are exported from the nucleus (3) and are further processed by the RNase Dicer (4). This releases the mature miR molecule and the miR*, the latter is degraded in most cases. Via partial sequence complementarity, the miRNA guides the RNA-induced silencing complex (RISC) to the 3′ untranslated region of protein-coding mRNAs (5). The RISC complex containing Argonaute 2 (Ago2) represses translation of mRNAs and may also reduce the abundance of the mRNA molecule. This leads to reduced protein abundance (6) and affects biological processes by targeting key molecules or by affecting groups of molecules that may be part of common regulatory processes (7). Furthermore miRNA molecules might be released from the cell and are detectable in the extracellular fluids and may exert effects on other types of cells (8). The expression of miRNAs is highly regulated not only at the level of transcription (12), but also posttranscriptional processing of the miRNA may be regulated (10). The effect of the miRNA on a specific target is modulated by many factors including abundance of the miRNA and target molecule in a specific cell, but potentially many other factors like localization of interacting molecules or other RNA-interacting molecules will influence miRNA function (9). Interestingly several cases have been identified where miRNAs regulate proteins that themselves may regulate the abundance of the miRNA (11), establishing stabilizing regulatory feedback loops
Of special importance for the binding of a miRNA to its target 3′ UTR is the seed sequence that is constituted by the bases 2–7 at the 5′end of the miRNA. Based on such findings, algorithms have been developed and successfully used to predict miRNA-mRNA interactions. Target prediction tools like TargetScan [5] use sequence complementarity of miRNAs to potential target mRNAs to predict the miRNA-mRNA interaction. In addition, conservation of binding sites across species is used to identify functionally important target sites; however, the algorithms predict hundreds to thousands of potential target sites per miRNA. In many cases these potential target sites can be validated in reporter assays; however, whether the expression of a target mRNA is regulated by a miRNA in vivo depends on more factors: for instance, other interacting molecules, expression level, and turnover of miRNA and target RNA. Therefore, under physiological conditions, only a few of the predictable target molecules are indeed regulated by the miRNA, and the functional significance of such regulatory events is not always obvious. Consequently, regulatory events mediated by miRNAs need to be investigated in models that closely resemble in vivo physiological conditions to identify relevant mechanisms.
The RNAse Dicer is essential for the maturation of microRNAs. Deletion of this key enzyme leads to depletion of mature miRNAs. In mice, germ line deletion of Dicer resulted in early embryonic lethality during gastrulation [6]. To circumvent this early lethality and to allow investigation of the requirement of miRNAs for cardiovascular development, other methods such as tissue- and time-specific deletion of Dicer using the loxP-Cre recombinase system were used to obtain conclusive results. Here, the Cre recombinase is expressed in specific tissues at defined developmental stages using appropriate promoters. The Cre recombinase deletes genomic sequences that have been engineered to be flanked by loxP sequences. Fusion of Cre recombinase to domains of the estrogen receptor can be used to induce Cre recombination upon application of tamoxifen to the animals. Cardiac-specific deletion of Dicer in early cardiac progenitors, using NKX2.5-driven Cre recombinase, leads to embryonic lethality due to heart failure 4 days after deletion of Dicer [7]. Deletion of Dicer at an early fetal stage of development using an α-myosin-heavy-chain-driven Cre recombinase resulted in dilation of the heart, and mutant mice died shortly after birth [8]. These Dicer knockout hearts displayed aberrant expression of cardiac contractile proteins, as well as sarcomeric disorganization.
The importance of miRNAs for heart development is further confirmed by the studies using cardiac- and skeletal muscle-specific deletion of DGCR8 in mice. Perinatal deletion of the RNA-binding protein DGCR8, which is part of the miR processing complex in the heart, resulted in postnatal lethality [9]. The importance of miRNA-mediated regulation for the development of VSMCs has been demonstrated by deletion of Dicer. Smooth muscle-specific deletion of these enzymes leads to extensive hemorrhage at embryonic stages and dilatation of thin-walled blood vessels, caused by a decrease in proliferation of VSMCs [10]. Deletion of Dicer in endothelial cells was compatible with survival of animals; however, angiogenic responses induced in postnatal animals were reduced [11].
In summary, it is clear that miRNAs are essential for cardiovascular development. Every type of cell in the cardiovascular system expresses numerous miRNAs, and many of these miRNAs are more broadly expressed and thus might fulfill more general functions. Thus, we need to focus on the analysis of specific miRNAs to identify specific functions of miRNAs in cardiovascular development (Table 62.1).
Table 62.1
Summary of important functions of miRNAs in cardiovascular development
microRNA | Type of cell | Function | Ref. |
|---|---|---|---|
miR-1/133a cluster | Cardiomyocyte | Regulation of smooth muscle gene expression, control of differentiation | |
miR-208/499 family | Cardiomyocyte | Regulation of structural identity, mediation of cardiomyocyte hypertrophic response | |
miR-15 family | Cardiomyocyte | Supports the developmental switch from cardiomyocyte proliferation to hypertrophic growth | |
miR-17–92 cluster | Cardiomyocyte | Control of cardiomyocyte proliferation | |
miR-16b–25 cluster | Cardiomyocyte | Control of cardiomyocyte proliferation, paralogous to miR-17–92, | [19] |
miR-92 | Endothelial cell | Limits angiogenesis | [21] |
miR-126 | Endothelial cell | Affects vascular integrity and angiogenesis, promotes endothelial cell proliferation after injury | |
miR-218 | Endothelial cells | Essential for endocardial and endothelial cell migration | |
miR-143/145 cluster | Vascular smooth muscle cells | Maintenance of contractility and contractile phenotype |
62.3 Specific Functions of Individual miRNAs in Heart Development
62.3.1 miR-1/133a Are Essential for Early Embryonic Heart Development
The microRNAs miR-1 and miR-133a are among the most abundant miRNAs found in cardiomyocytes and are specifically expressed in heart and skeletal muscles (Chap. 13). Both miRNAs are evolutionary conserved and are encoded in two miR-1/133a clusters in the mammalian genome. In humans the miR-1-1/miR-133a-2 cluster is found in an intron of a protein-coding gene on chromosome 20. The miR-1-2/miR-133a-1 cluster is located intergenically on chromosome 18. Both clusters are almost equally expressed in cardiomyocytes and give rise to identical mature miR-1 and miR-133a molecules. The expression of these miRNAs is controlled by factors including SRF (serum response factor), MEF2 (myocyte enhancer factor 2), and MyoD that also induce expression of other muscle-specific genes [28, 29].
Different loss-of-function studies demonstrated that the miRNAs miR-1 and miR-133a are essential for proper cardiac development. Deletion of miR-1-2 resulted in a partially penetrant phenotype with ventricular septal defects (VSD), and this was attributed to upregulation of Hand2 transcription factor as a direct target of miR-1 [7]. Deletion of single miR-133a copies did not cause any obvious phenotype, but the complete deletion of both miR-133a genes also resulted in a partial penetrant phenotype with VSD, increased proliferation of neonatal cardiomyocytes, and increased expression of smooth muscle genes in the heart. These observations after complete loss of miR-133a were attributed in part to the upregulation of the direct miR-133a targets cyclinD2 and SRF [12]. In contrast to the previous studies, deletion of both miR-1/133a clusters (dKO) leads to massive defects in early cardiac development [13]. Whereas the initial cardiac specification is not disturbed, deletion of both miR-1/133a clusters leads to massive upregulation of smooth muscle markers in the embryonic heart, reduced proliferation, and thinning of the compact layer of the myocardium.
The miR-1 target myocardin and the miR-133a target Kcnmb1 (calcium-activated potassium channel subunit beta-1) were strongly upregulated after loss of miR-1/133a. Transgenic overexpression of myocardin in the embryonic heart reproduced many aspects of the miR-1/133a phenotype including upregulation of smooth muscle markers, proving that indeed myocardin is an essential miR-1 target that needs to be repressed by miR-1 for proper cardiac development. Myocardin is a cofactor of SRF, and thus increased expression of myocardin also leads to upregulation of miR-1/133a expression. Accordingly, miR-1 and myocardin constitute a regulatory feedback loop that also controls expression of miR-133a and its targets (Fig. 62.2a). Myocardin additionally activates transcription of the smooth muscle-specific potassium channel Kcnmb1 that in turn is a direct target of miR-133a. Taken together these results establish that the miR-1/133a clusters are not needed for initial specification of cardiac cells but act as functional units to repress smooth muscle markers. Moreover, after deletion of miR-1/133a in the embryonic heart, additional previously identified miR-1/133a targets like SRF, Hand2, or Irx5 were not changed in their expression, indicating that regulation of targets might also depend on the developmental stage of the cells or on other modifying factors. These findings were confirmed by the recent miR-1-1 knockout that had a similar phenotype like the miR-1-2 mutants; however, the phenotype also was influenced by the genetic background of mice. Mutation of both miR-1 copies resulted in postnatal lethality due to severe dysfunction and disturbed sarcomeric structures [14]. Although the miR-1 mutant mice survive until 2 weeks after birth, the upregulation of myocardin is the likely cause for the observed ectopic smooth muscle gene expression in the heart.
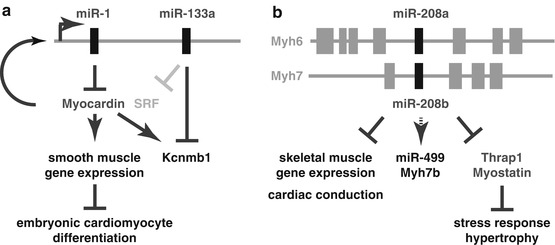
Fig. 62.2
Functions of miRNAs in cardiomyocyte development. miRNAs affect development of the heart at different stages. (a) Aside from other functions at later developmental stages, the miRNA cluster miR-1/133a is essential to modulate smooth muscle gene expression in the early heart. In early heart development, well before cardiac chamber formation, cardiomyocytes (CMs) express several smooth muscle marker genes that are repressed during later stages of development. At embryonic stages, miR-1/133a is essential to repress myocardin that together with SRF drives the expression of smooth muscle genes, including Kcnmb1. In a feedback loop, myocardin drives expression of its repressor miR-1/133a, finally leading to repression of smooth muscle gene expression during further cardiomyocyte development. miR-133a directly represses smooth muscle-specific genes like the potassium channel Kcnmb1. In adult stages, miR-133a additionally represses translation of SRF to repress smooth muscle gene expression in adult CMs. (b) miRNAs of the miR-208 family have functions in postnatal development of the heart. miR-208a/b are encoded in the myosin-coding genes Myh6/7 and repress the expression of skeletal muscle-specific genes in the heart. Moreover, the miRNAs drive expression of miR-499 and of its host gene Myh7b and are essential to support stress responses of the heart like the hypertrophic growth after specific challenges
62.3.2 miRNAs Affecting Proliferation and Morphogenesis of the Heart
The conserved miR-17–92 cluster comprises six microRNAs (miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, miR-92-1) that are processed from one precursor transcript [19]. Many functions have been assigned to this cluster such as oncogenic potential, functions in skeletal development, and B-cell differentiation. In addition to this cluster two paralogous clusters are found in the genome: the miR-106a–363 (miR-106a, miR-18b, miR-20b, miR-19b-2, miR-92-2, -363) and the miR-106b–25 cluster (miR-106b, miR-93, miR-25). Based on identical seed sequences, the miRNAs in these clusters can be grouped into four miRNA families suggesting at least partial redundancy of the clusters. The miRNA clusters also have a role in cardiovascular development as miR-17–92-deficient mice die after birth due to lung hypoplasia and ventricular septal defects (VSDs), whereas mice deficient of miR-106a–363 or miR-106b–25 cluster are viable. Concomitant deletion of miR-17–92 and miR-106b–25 clusters was embryonically lethal prior to embryonic day 15 with severe disturbance of heart development with thinner ventricular wall and defective ventricular and atrial septation [19]. Heart-specific deletion and overexpression of the miR-17–92 cluster demonstrate that the cluster is required and sufficient for CM proliferation in postnatal and adult hearts [20].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


