Figure 9–1.
Conceptual scheme of cardiac electro-mechanical regulation. Electrical control of cardiac contraction is via Excitation-Contraction Coupling (ECC), while the mechanical environment affects cardiac electrical behaviour via Mechano-Electric Feedback (MEF) (From Kohl et al. [1]. Reprinted with kind permission from Elsevier)
Spatio-Temporal Considerations
The heart is a spatially heterogeneous organ, both in terms of structure (e.g. variations in regional tissue architecture, cell distribution, coupling, innervation, or blood supply) and function (active and passive tissue electro-mechanical properties). In addition, there are temporal gradients in key electro-mechanical behaviour (e.g. activation, repolarization, contraction, relaxation). Nonetheless, under physiological conditions the heart functions as a highly coordinated unit. This ‘externally homogeneous’ mechanical performance at the organ level arises from, and necessarily requires, structural and functional heterogeneity, from sub-cellular to whole organ levels [2].
While this appreciation has guided our thinking on structural and functional adaptation of the heart to (patho-)physiological developments, it leaves the question as to how an individual cardiomyocyte inside this complex organization ‘knows’ when and how to respond to beat-by-beat changes in electro-mechanical activity. The when is largely a function of the coordinated spread of electrical excitation. The how requires sub-cellular regulatory pathways that match local mechanical performance to global mechanical demand; underlying processes are discussed next.
Excitation Contraction Coupling
Introduction
The process of ECC involves a transient rise in free cytosolic calcium concentration ([Ca2+]i), as the intermediate signal between electrical depolarization of the cell membrane and activation of contractile myofilaments. The sequence is initiated by calcium (Ca2+) influx across the sarcolemma, which produces a secondary release of Ca2+ from the sarcoplasmic reticulum (SR, a specialized Ca2+-storage compartment in mammalian cardiac myocytes). The SR can rapidly release Ca2+ in response to sarcolemmal Ca2+-influx, a process termed ‘Ca2+-induced Ca2+-release’. The released Ca2+ then binds to several intracellular Ca2+-binding proteins, including troponin, which in turn activates the myofilaments. The majority of Ca2+ is re-sequestered back into the SR during each heartbeat, while the remainder (in steady-state this is an amount equivalent to the initial trans-sarcolemmal influx) is extruded across the membrane.
In reality, the events involved in ECC are intimately connected and cannot be decomposed into discrete sets of spatially or temporally defined stages. For example, SR Ca2+-release occurs simultaneously with re-uptake, with the net effect depending on the relative balance of the competing processes at any given time. Also, SR Ca2+-storage involves ‘memory’ mechanisms that render the amount of Ca2+ stored in, and released from, the SR strongly dependent on stimulation history. Thus, the system shows complex behavior within single beats and between multiple beats. The following sections outline separate steps in the activation and relaxation sequence of cardiac muscle. However, care must be taken to remember that this decomposition into discrete steps is highly artificial and underestimates the complex dynamics of ECC [3, 4].
From Action Potential to Calcium Release
Figure 9.2 shows a conceptual model of Ca2+-handling during a typical heartbeat. The trigger events (Fig. 9.2a) involve sarcolemmal influx of Ca2+ through L-type Ca2+ channels (LCC; thick downward arrow), which produces a secondary and larger release of Ca2+ from the SR (upward thick arrow), released via ryanodine receptors (RyR) into the diadic space (dotted box labeled DS). An important negative feedback pathway exists in that the released Ca2+, as well LCC influx itself, cause Ca2+-induced inactivation of LCC, which reduces further influx (upward thin arrow with ‘minus’ sign). In addition, the cytoplasmic Ca2+ level can also produce inactivation of LLC. From the diadic space, Ca2+ diffuses into the myoplasm (thick rightward arrow) to increase [Ca2+]i from approximately 0.1 to 1 μM (represented by the schematic Ca2+-transient). The majority of this released Ca2+ binds to intracellular buffers, including troponin and calmodulin (not shown). Ca2+ can also enter the cell during the action potential (AP) upstroke and subsequent ‘notch’ (rapid partial repolarisation prior to the AP plateau) via the Na+/Ca2+ exchanger (NCX), although this pathway is considered less important for ECC than Ca2+-influx via LCC.
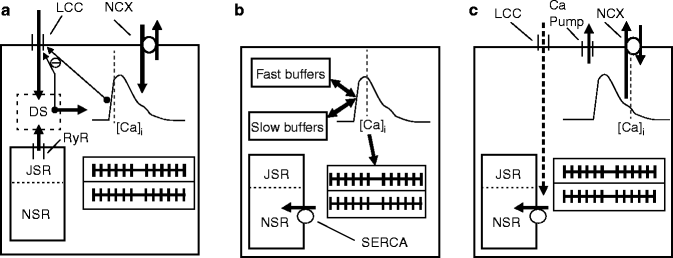
Figure 9–2.
Schematic diagram of a cardiac cell, showing three conceptual steps in excitation-contraction coupling (ECC), with pseudo-representative timing relative to cytosolic free calcium-concentration ([Ca2+]i) dynamics indicated by grey dotted lines. (a) Ca2+-induced Ca2+-release; (b) Cytosolic Ca2+-buffering and contraction changes; (c) Ca2+-reuptake and -extrusion (for detail see text). DS dyadic space, JSR junctional sarcoplasmic reticulum, LCC L-type Ca2+ channel, NCX Na+/Ca2+ exchanger, NSR network sarcoplasmic reticulum, RyR ryanodine receptor, SERCA sarcoplasmic/endoplasmatic reticulum Ca2+-ATPase
L-Type Ca2+ Channel Influx
The LCC is the major influx pathway for Ca2+ during each heartbeat. This current has multiple roles in producing the Ca2+-transient, both by directly increasing [Ca2+]i, and by triggering a larger secondary Ca2+-release from the SR. Moreover this current contributes to AP morphology, especially in sustaining the AP plateau in spite of repolarizing K+ currents. LCCs are activated by voltage and inactivated by both voltage and Ca2+, but of the two, Ca2+-based inactivation predominates in determining the amplitude and time-course of LCC currents [5, 6]. One potential role of this negative feedback (Fig. 9.2a, thin arrow) is to limit Ca2+-influx after triggering Ca2+-induced Ca2+-release. However, longer-term roles in Ca2+-homeostasis of both intracellular and SR Ca2+-levels are also proposed [4]. Interestingly the LLC inactivates in response to both dyadic space Ca2+ and cytoplasmic Ca2+ (as shown schematically in Fig. 9.2), in spite of favored physical proximity and higher amplitude of the dyadic space Ca2+ signal. Recent findings at the molecular level have elucidated a temporally based mechanism that can separately sense both the high amplitude, Ca2+ impulses in dyadic space and the low amplitude, prolonged Ca2+ levels in the cytoplasm [6–8]. The Ca2+-based inaction involves molecular interactions of the channel with a constitutively bound calmodulin, whose genetic manipulation to remove the negative feedback has surprisingly large effects on AP duration (lengthening it by four to five times) [5]. Thus, key electrophysiological parameters can be significantly more sensitive to Ca2+-handling than customarily assumed.
SR Ca2+ Release via RyR Channels
Ca2+-release from the SR via RyR has been shown to be roughly proportional to the trigger influx of Ca2+ via LCC, although some variation exists, depending on how Ca2+-flux is determined [8]. The system shows high gain, in that a small number of LLC will trigger release from a nearby cluster of RyR channels that open synchronously. As the discrete opening of LCC can modulate the number of activated RyR clusters, the large total Ca2+-release tracks roughly linearly with total LCC Ca2+-influx.
Another important property is that SR Ca2+-release is a nonlinear, steeply increasing function of SR Ca2+-load. Essentially no SR Ca2+-release occurs below a threshold, after which SR rises steeply with SR load [8, 9]. Interestingly, there are indications that more than 100 % of the original SR Ca2+-content can be released, suggesting that Ca2+ is recycled back into SR during the release process, and re-released again [8]. For a typical beat, though, Ca2+-release fraction is estimated at 60–80 % [9]. Interestingly, there appears to be a set maximum SR Ca2+-load beyond which spontaneous RyR Ca2+-release occurs, with potential for arrhythmogenesis. In addition, spontaneous RyR Ca2+-release appears to also play critical roles in determining resting [Ca2+]i.
From Calcium Release to Contraction
The translation of [Ca2+]i to force generation occurs at the level of the sarcomere, i.e. the basic subcellular unit of the contractile apparatus (Fig. 9.3). The sarcomere is composed primarily of interdigitated thick myosin filaments that interact, when activated, with thinner actin filaments (for reviews see [11, 12]).
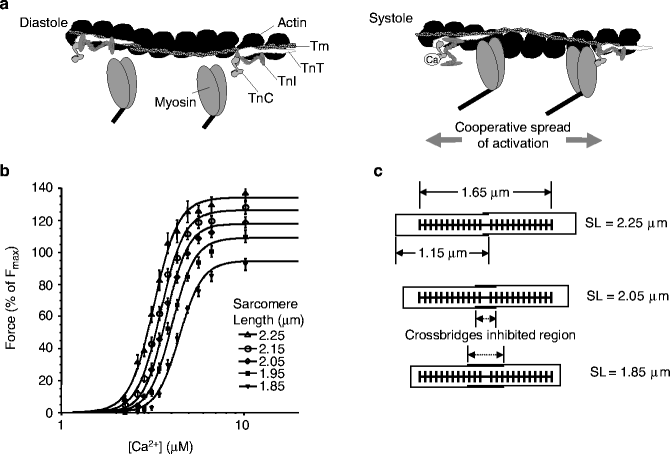
Figure 9–3.
Contractile filaments and calcium. (a) Interrelation of thin actin filament with nearby myosin heads. During diastole (left), tropomyosin (Tm) and troponin subunits (TnC, TnT, and TnI) sterically block crossbridge formation. During systole, elevated [Ca2+]i shifts the locations of tropomyosin/troponin to allow myosin head attachment and force generation. Note that cooperative activation can spread along the thin filament (see text for details) (From Bers [3]. Reprinted with permission from Springer). (b) Average force-Ca2+ relationships (pooled data, skinned rat cardiac trabeculae, n = 10) at five sarcomere lengths (SL); data are fitted to a Hill relationship. The level of cooperativity, assessed by Hill coefficient, is not affected by SL (From Dobesh et al. [10]. Reprinted with kind permission from The American Physiological Society). (c) Diagram showing expected sarcomere geometry for three of the SL shown. Thick filaments are schematically shown by the thick lines with protrusions to represent the myosin heads. Thin filaments are shown as horizontal thin lines that emanate from Z-disks, represented by the vertical lines. SL is defined as the spacing between two neighbouring Z-disks. Binding of crossbridges is assumed to be inhibited in the central region in which the thin filaments overlap, and hence the SL determines the degree of thin filament overlap and, ultimately, the number of recruitable crossbridges
While the basic interactions in the sarcomere are known, the underlying basis of several complex behavioral properties is still debated. For example, the myofilament system shows a high level of Ca2+-sensitivity, as characterized by steep Force-Ca2+ relationships with a Hill coefficient equal to seven or more (Fig. 9.3b). Because each troponin binds a single Ca2+ ion on the regulatory site of Troponin C (TnC), a Hill coefficient of one is predicted; hence, the high Ca2+-sensitivity is assumed to result from one or more cooperative mechanisms (for review see [13]).
Briefly, three types of cooperative interactions are most widely accepted. Attached crossbridges have been shown to increase the Ca2+-affinity of TnC [14–16]. A second type of cooperativity among the regulatory proteins is thought to arise from nearest-neighbor interactions, produced by the overlap of adjacent tropomyosin units along the thin filament (Fig. 9.3a) [11, 12]. A third proposed mechanism is that the binding of one myosin head increases the binding rate of neighboring heads by holding the regulatory proteins in a more permissive conformation [17]. Alternatively, the binding of one crossbridge may pull binding sites on a compliant thin filament into register with myosin heads that have an inherently different characteristic distance in their repeating structure [18].
Another complex phenomenon is the length-dependence of force-Ca2+ functions, often studied in isometric conditions (i.e. length is prescribed, while force is measured). Length-dependent effects can be separated into two main categories: changes in plateau force and changes in Ca2+-sensitivity. As SL increases, changes in maximum Ca2+-activated force may arise from changes in overlap of thin filaments, that increase the recruitable pool of crossbridges (Fig. 9.3c; note that the lengths of the thick and thin filaments differ from the accepted values for skeletal muscle [13, 19]). In support of this scheme, developed force and ATPase rate of maximally activated cardiac muscle are linear functions of SL [20]. The second category is a decrease in myofilament Ca2+-sensitivity, as shown by the leftward shift in force-Ca2+ curves as SL increases (Fig. 9.3b). These length-dependent changes in Ca2+-sensitivity are generally assumed to be the cellular basis of the Frank-Starling effect, although the biophysical basis is still under debate.
Another hypothesis suggests that SL changes alter the lattice spacing of the thick and thin filaments because the isovolumetric nature of cardiac cells produces a narrowing of cross-section with increasing cell length [21, 22]. Titin is a giant, infolded protein that runs the length of the thick filament and then connects across to the root of thin filaments and on to the lattice of proteins that comprise the Z-disc. Titin is ideally positioned, therefore, to sense SL (while titin is not shown in Fig. 9.3c, the span from the thick filament to the Z-disk distance is clearly a function of SL for the range shown). Indeed, selective removal of titin by mild treatment with trypsin (0.31 μg/mL for 10–15 min) affects both myofilament Ca2+ sensitivity and inter-filament spacing. Titin could modify crossbridge binding by modifying interfilament spacing [23], or could act by other mechanisms such as changing the angle that myosin heads emanate from the thick filament, a feature speculated to produce SL-dependent changes in Ca2+ sensitivity [22, 24]. In these schemes, the local concentration of myosin heads in the vicinity of the thin filament will modulate effective binding and cycling rates of crossbridges, ultimately modifying Ca2+-sensitivity and developed force.
The inter-filament spacing hypothesis was proposed in the early 1980s [22] and is supported by several more recent reports [25]. However, other studies suggest that physiological changes in lattice spacing are insufficient to account for the observed changes in Ca2+-sensitivity (for review see [26]). In addition to inter-filament spacing and titin effects, several other mechanisms have been proposed to explain the length-dependence of Ca2+-sensitivity. Some mathematical models suggest changes in Ca2+-sensitivity could result from cross-interactions of cooperative mechanisms with length-dependent changes in crossbridge recruitment [13, 19]. Clearly, more work is needed to elucidate the mechanism of this fundamental property of cardiac myofilament mechanics.
In addition to the immediate stretch-induced increase in cardiac force development, there is a secondary and more slowly occurring increase in force, which eventually reaches a plateau [27]. In contrast to the fast response, the slow force response involves an increase in Ca2+-transient amplitude (Fig. 9.4) [28]. At the level of the myofilaments, there are no slow time-dependent changes in Ca2+-sensitivity. In fact, length changes during the diastolic period alone are sufficient to generate a slow force response [29, 30], a finding that rules out the myofilament-based mechanisms discussed above. A full review of slow changes in force, and possible effects on membrane currents, is available elsewhere [31]. One obvious source of increased Ca2+ are mechanically modulated ion channels (discussed below), which either directly conduct Ca2+ ions, or allow Na+ entry, which secondarily increases intracellular Ca2+, for example via NCX.
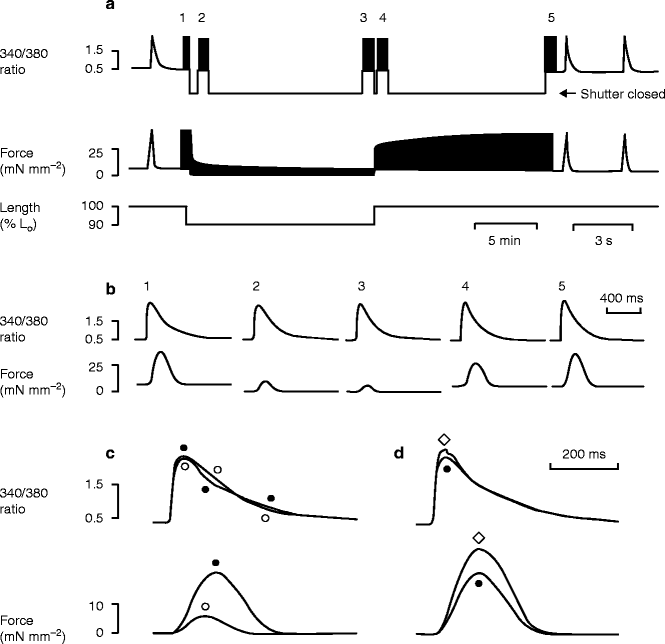
Figure 9–4.
Illustration of immediate and slow changes in force after length changes. Records of the changes in fura2 fluorescence ratio and force produced by shortening a rat trabecula by 10 % for 15 min. (a) Records of 340 nm/380 nm fluorescence ratio and force, with a representation of the length change from the initial length (L0). A shutter in the excitation light pathway was opened only for discrete 48 s recording periods (labelled 1–5) in order to avoid photobleaching of fura2. Note the slow changes in twitch force after the changes in muscle length. (b) Mean records (from 16 twitches) of fluorescence ratio and force measured during periods 1–5 in a. (c) Overlaid traces of the fluorescence ratio and force averaged during periods 3 (○) and 4 (●) to illustrate the rapid effects of the length increase. Resting forces have been subtracted from these traces. (d) Similar overlaid traces averaged during periods 4 (●) and 5 (◊) to illustrate the delayed effects of length increase. 24 °C, 1 mM external Ca2+, 0.33 Hz stimulation rate (From Kentish and Wrzosek [28]. Reprinted with kind permission from John Wiley and Sons)
More recently, contributions by second messenger systems have been proposed. Only a brief description is provided here, as more detailed reviews are available elsewhere [32]. It has been proposed that Na+ influx occurs via the Na+/H+ exchanger (NHX) that may be activated by a stretch-induced increase in angiotensin II (AT-II) and endothelin-1 (ET-1) [33, 34]. More recently, several groups have proposed intermediate steps from AT-II/ET-1 activation to NCX. One pathway includes the generation of reactive oxidative species, which activate a cascade including extracellular signal-regulated kinases 1 and 2 and p90 ribosomal S6 kinase. These eventually lead to phosphorylation and activation of NHX [35]. An alternative pathways has intermediates that include phospholipase C and protein kinase C [36, 37], or cyclic adenosine monophosphate and protein kinase A [38]. Another proposed mechanism [39] involves generation of reactive oxidative species that, in turn, activate phospholipases C and A2, which generate amphipaths (diacyl glycerol and arachidonic acid, respectively). Amphipaths are thought to modify the local curvature of the membrane, and this can activate membrane-bound channels, including the transient receptor potential cation channel member 6 (TRPC-6), a cation non-specific stretch activated channel [40]. A complicated picture is emerging in which the second messenger systems are complex, perhaps partially redundant, and with the potential for substantial crosstalk between biophysical and biochemical properties of the cell.
Effects on the Ca2+ Transient
While the exact mechanisms of length-dependent changes in Ca2+-sensitivity are controversial, they may not be that important to understanding mechanical feedback on Ca2+-transients. The amount of Ca2+ bound to the myofilaments is generally assumed to be affected by the number of attached crossbridges (instead of length itself) [14–16]. Hence, any feature that affects developed force can have secondary effects on Ca2+-binding to troponin, and on the Ca2+-transient.
For example, increasing muscle length by 10 % (see time-points 3 and 4 in Fig. 9.4) has a dramatic effect on developed force (see Force in Fig. 9.4b). This increase in developed force leads to greater Ca2+-binding to TnC, initially lowering cytosolic [Ca2+]i. Later, during the Ca2+-transient, [Ca2+]i is slightly higher, as more Ca2+ is slowing coming off TnC (compared with the low force case). Similar effects on Ca2+-transient have been seen elsewhere [14, 28, 29, 41, 42], although details depend on the nature of mechanical perturbations and Ca2+-monitoring agents used. Taken together, the results suggest that changes in force-dependent Ca2+-binding to troponin have a noticeable, but not dramatic, effect on [Ca2+]i levels, which may restrict the role Ca2+-buffering in mediating MEC effects in normal tissue. However, more profound MEC effects, leading to arrhythmic behavior, have been reported for pathological conditions, especially at the border between regions of strong and weak contraction, both in single cells [43] and trabeculae [44] (see section “Systolic Stretch Effects”).
Finally, a few more general comments should be made about Ca2+-buffering. The cytosol of the cardiomyocyte is heavily buffered, and the vast majority of Ca2+ ions are bound to several important buffers. For example, at diastolic [Ca2+]i of 0.1 μM, only ∼2.5 % of Ca2+ ions are ‘free’ in solution; this fraction increase to ∼4.5 % when systolic [Ca2+]i rises to 1 μM [3]. As shown in Fig. 9.3b, fast and slow buffering systems exist in addition to troponin and the SR. Several fast buffers have dissociation constants near the operating range of the Ca2+-transient (0.1–1 μM), and these bind and release Ca2+ on each heart beat. This group includes calmodulin, adenosine tri-phosphate (ATP), creatine phosphate, and the phospholipids of the sarcolemma. Slow buffers have dissociation constants that are much lower than the operating range of [Ca2+]i, and hence Ca2+ remains largely bound to these buffers throughout the heartbeat. Slow buffers include myosin and two high-affinity, non-regulatory sites on TnC. In addition, the mitochondria comprise roughly 35 % of cytosolic volume and can potentially contain large amounts of Ca2+. Their large volume, coupled with close proximity to myofilaments and RyR, suggest a potentially significant active role for mitochondria in ECC [3]. However, most findings suggest little net Ca2+ transit between mitochondria and cytosol on a beat-by-bet basis. In fact, mitochondrial Ca2+-exchange may be an epiphenomenon of mechanisms that match ATP production to cellular consumption, using intracellular Ca2+ as a proxy for the energetic demand of a cell.
From Contraction to Relaxation
While ‘contraction’ tends to be at the focus of attention, the process of relaxation is just as important for cardiac performance, yet less well understood [45]. As diastolic filling occurs under low pressure, any residual force from incomplete relaxation can severely affect cardiac function. Relaxation is more than just elastic recoil of tissue, and complexities arise from the interplay of Ca2+-release from the myofilaments, Ca2+-reuptake into the SR, and crossbridge detachment (Fig. 9.2c).
Besides serving as a trigger, LCC Ca2+-influx also serves to load the SR (Fig. 9.2c, dotted down-arrow). This influx of Ca2+ is thought mainly to influence the amount of Ca2+ releasable by the SR on the subsequent beat (rather than the current beat). In steady-sate conditions, net loading is zero, as the amount of Ca2+ extruded by NCX matches that of LCC influx.
The majority of [Ca2+]i (60–80 %, depending on species) is recycled back into SR via the ‘sarcoplasmic/endoplasmic reticulum Ca2+-ATPase’ (SERCA). Factors such as heart rate, inotrophic stimulation, or pathologies affect re-uptake rates (heart failure can decrease uptake by 50 %) [3, 46]. The major sarcolemmal efflux pathway is the NCX, extruding ∼20–40 % of Ca2+ during the transient, whereas the sarcolemmal Ca2+-ATPase and mitochondrial uptake are generally thought to contribute less [47].
With the decline of [Ca2+]i, Ca2+ ions unbind from TnC, starting the relaxation process. If TnC were a simple buffer, the process could be easily described. However, complexities arise from the presence of activated thin filaments and attached crossbridges, which increase TnC affinity for Ca2+ (as described previously in the section “From Calcium Release to Contraction”). Moreover, a small number of attached crossbridges are thought to be able to hold the thin filament in an activated state, even if Ca2+ has dissociated from neighbouring binding sites (Fig. 9.3a). Thus, attached crossbridges can slow relaxation both by increasing Ca2+-affinity of TnC, and via holding the thin filament activated after Ca2+ has dissociated from a fraction of TnC.
Because of these features, contractions involving high levels of developed force (and many attached crossbridges) are slower to relax than those with lower force development. This affects especially final relaxation, past 50 % of maximum force [48, 49]. Thus, a larger force transient as is observed after application of stretch (see Fig. 9.4c, lower panel) is noticeably slower to relax than a low force transient (note that for the two runs shown in Fig. 9.4c, the activating Ca2+-transients are very similar). Interestingly, increased force due to larger peak Ca2+-transients also produces a slowed final relaxation, suggesting that the effect results from slow unbinding kinetics of crossbridges, not upstream Ca2+-activation events [45, 49].
Finally, and as noted previously, complete relaxation is important for competent diastolic filling. Here, the steep Ca2+-sensitivity of the myofilaments contributes to keeping developed force minimal at diastolic [Ca2+]i. Other features that may promote relaxation include the ability of myosin to detach under isometric or lengthening conditions (by quickly reversing the steps of attachment and head rotation) in a manner that retains most of the energy of ATP hydrolysis [13, 45]. In contrast, the typical forward cycling scheme requires ADP dissociation and new ATP binding for a crossbridge to detach (if only this slower detachment mechanism existed, relaxation rate would be greatly slowed). In fact, recent characterizations of isolated myofibrils suggest that relaxation is a non-uniform biphasic process [45], and while isolated myofibril behaviour may differ from intact muscle, the point is clear that relaxation is complex and is not just the reverse of activation.
Mechano-Electric Coupling
Diastolic Stretch Effects
As shown above, the heart is an exquisitely mechano-sensitive organ. This also applies to mechano-electric transduction, as will be obvious to anyone whose training involved the use of Langendorff-style heart preparations that can be stopped or started ‘by the flick of a finger’. Mechanisms underlying these acute electrical responses to mechanical stimulation include stretch-activated ion channels (SAC), mechanical modulation of Ca2+-handling, and effects mediated via communication with other cells.
As a rule, mechanical stimulation of resting cardiac tissue (during ‘electrical diastole’) causes membrane depolarization (Fig. 9.5) [50]. This behavior can be observed across the whole range of structural complexity, from whole organ to single cell, and it is understood to be largely mediated by depolarizing trans-membrane currents through SAC [51, 52].
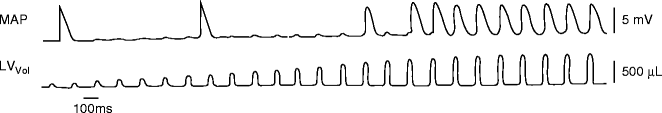
Figure 9–5.
Diastolic stretch causes membrane depolarization. Monophasic action potential recordings (MAP) from an isolated rabbit heart (AV node disrupted) shows that injection of increasing amounts of fluid into a left-ventricular balloon (LV Vol ) causes matching diastolic depolarisations which, once threshold is reached, will mechanically pace the preparation (note: the first two action potentials are spontaneous escape beats not related to mechanical stimulation) (From Franz et al. [50]. Reprinted with kind permission from Wolters Kluwer Health)
SAC have been discovered in mammalian cardiomyocytes 25 years ago [53], and a number of channels with varying ion selectivity and gating properties have subsequently been described. Despite over two decades of research, a uniform SAC classification has yet to emerge.
Meanwhile, SAC can be distinguished based on ion selectivity. SAC that show little selectivity for the various cations normally present in physiological solutions comprise the first group (SACNS; for Non-Selective). The second group preferentially conducts potassium (SACK). A third group of chloride-selective channels [54] has been described in settings that involve centrifugal membrane deformation (most commonly in the context of patho-physiologically increased cell volumes). The latter channels show significant lag-times between mechanical stimulus and increased channel open probability (1 min or more), rendering them less likely to underlie acute mechanical effects on heart rate and rhythm. (Mechano-sensitive chloride channels are likely to affect electrophysiology predominantly in settings such as ischaemia, reperfusion, or hypertrophy, where they are constituently activated; for further detail see [41]).
SACNS usually have reversal potentials between 0 and −20 mV. This is more positive than the membrane potential of resting cardiomyocytes, so that diastolic activation of SACNS will tend to depolarize cardiac cells. In working cardiomyocytes, SACNS activation may cause ectopic AP generation [51], while in pacemaker cells a positive chronotropic response can be observed [55].
SACK activation will tend to shift the membrane potential towards the potassium equilibrium potential (negative to –90 mV), so their activation will oppose depolarization, or aid repolarization.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


