Lionel H. Opie, Donald M. Bers The major function of cardiac muscle cells (cardiomyocytes or myocytes) is to execute the cardiac contraction-relaxation cycle. The contractile proteins of the heart lie within these myocytes, which constitute approximately 75% of the total volume of the myocardium, although only approximately one third of the number of all the cells.1–4 Approximately half of each ventricular cell is occupied by the myofibrils of the myofibers (Fig. 21-1) and approximately one quarter to one third by mitochondria (Table 21-1). A myofiber is a group of myocytes (Fig. 21-1) held together by surrounding collagen connective tissue, the latter being a major component of the extracellular matrix. Further strands of collagen connect myofibers to each other. The individual contractile myocytes account for more than half the heart’s weight. Ventricular myocytes are roughly brick shaped, typically 150 × 20 × 12 µm (Table 21-1), and are connected at the long ends (Fig. 21-2). Those in the atrium are smaller and more spindle shaped (<10 µm in diameter and <100 µm in length). When examined under a light microscope, atrial and ventricular myocytes have cross striations and are often branched. Each myocyte is bounded by a complex cell membrane, the sarcolemma (sarco = flesh; lemma = thin husk), and is filled with rodlike bundles of myofibrils (see Fig. 21-1), which are the contractile elements. The myocyte sarcolemma invaginates to form an extensive tubular network (the T tubules) that extends the extracellular space into the interior of the cell (see Figs. 21-1 and 21-2). Ventricular myocytes are typically binucleate, and these nuclei contain most of the cell’s genetic information. Some myocytes have one or several nuclei. Rows of mitochondria are located between the myofibrils and also immediately beneath the sarcolemma. Mitochondria function mainly to generate the energy, in the form of adenosine triphosphate (ATP), that is needed to maintain the heart’s contractile function and the associated ion gradients. The sarcoplasmic reticulum (SR) is a specialized form of endoplasmic reticulum that is critical for calcium (Ca2+) cycling, which is the on-off switch for contraction (see Fig. 21-1). When the wave of electrical excitation reaches the T tubules, voltage-gated Ca2+ channels open to provide relatively small entry of Ca2+, which triggers additional release of Ca2+ from the SR via closely apposed release channels. This is the Ca2+ that initiates myocardial contraction. Ca2+ sequestration by the SR causes relaxation (diastole). Anatomically, the SR is a lipid membrane–bounded, fine interconnected network spreading throughout the myocytes. The Ca2+ release channels (or ryanodine receptors [RyRs]) are concentrated at the part of the SR that is in very close apposition to the T tubular Ca2+ channel. These are called terminal cisternae (boxes or baskets, Latin) or the junctional SR (jSR). The second part of the SR, the longitudinal, free, or network SR, consists of ramifying tubules that surround the myofilaments (see Fig. 21-1) that take Ca2+ back up into the SR and thus drive relaxation. Such uptake is achieved by the ATP-requiring Ca2+ pump known as SERCA (sarcoendoplasmic reticulum Ca2+-adenosine triphosphatase [ATPase]). The Ca2+ taken up into the SR is then stored at high concentration, in part bound to storage proteins, including calsequestrin, before being released again in response to the next wave of depolarization. Cytoplasm or sarcoplasm refers to the intracellular fluid and proteins therein, but excludes the contents of organelles such as the mitochondria, nucleus, and SR. The cytoplasm is crowded with myofilaments, but this is the fluid within which the concentration of Ca2+ rises and falls to cause cardiac contraction and relaxation. The molecular signal systems that convey messages from surface receptors to intracellular organelles may be directed to specific sites by molecules that “anchor” components of the signaling cascades to specific loci, such as around beta-adrenergic receptors and Ca2+ channels at the T tubule–SR junction and caveolae (small flask-shaped sarcolemmal invaginations). Scaffolding proteins such as caveolin or the RyR bring interacting molecules closely together at these locations. These complexes can release components that translocate and signal elsewhere in the cell, such as the nucleus, where it can signal for myocyte growth. Another type of subcellular shuttling is involved in getting the ATP produced in mitochondria to sites where it is used (e.g., myofilaments), which is facilitated by the location of creatine kinase, an enzyme that converts creatine phosphate to ATP. The typical ventricular myocyte has approximately 8000 mitochondria, each of which is ovate with a long axis measuring 1 to 2 µm and short axis of 300 to 500 nm. There are two membranes, the outer and inner mitochondrial membranes (OMM and IMM; Fig. 21-3). The IMM is “crumpled” such that it forms a large surface area within a small volume, and it contains the cytochrome complexes that make up the respiratory chain, including F0-F1 ATP synthase. The space within the IMM, the mitochondrial matrix, contains enzymes of the tricarboxylic acid (TCA) cycle and other key metabolic components. These components provide reducing equivalent protons that are pumped out of the matrix by the cytochromes, and it is this proton pumping that creates the very negative matrix potential with respect to cytosol (Ψm = −180 mV). Such a negative Ψm creates a strong electrochemical gradient for protons, which as they flow down this energy gradient on F0-F1 ATP synthase, are responsible for making ATP. The ATP still needs to get out of the mitochondria, and an adenine nucleotide transporter exchanges mitochondrial ATP for cytosolic adenosine diphosphate (ADP). This system is exquisitely regulated to maintain cytosolic [ATP] and [ADP] constant during the dramatic changes in cardiac workload.5 The multiple control mechanisms involved in this process are not fully understood, but one is quite relevant to the excitation-contraction coupling process. Increased cardiac work in a physiologic setting is usually driven by higher-amplitude and/or more-frequent Ca2+ transients. This elevation in average [Ca2+]i also increases mitochondrial matrix [Ca] ([Ca2+]m), which activates key dehydrogenases in the TCA cycle and also pyruvate dehydrogenase to restore levels of nicotinamide adenine dinucleotide, reduced form (NADH), and thereby increase cytochrome activity and hence restore or maintain [ATP] toward normal levels. The above raises the issue of how mitochondria regulate [Ca2+]m because there is also a huge electrochemical gradient favoring entry of Ca2+ into mitochondria.3 Indeed, [Ca2+]m is normally not much different from [Ca2+]i and is kept at that level by a mitochondrial Na/Ca exchanger (NCXL), which uses the also steep Na+ electrochemical gradient to pump Ca2+ out of the mitochondria. However, this would of course load the mitochondria with Na+, so Na+ must also be extruded from the mitochondria. This is accomplished by an Na/H exchanger in the IMM, but a consequence is that this influx of H+ costs energy. That is, these protons could have entered the mitochondria via the F0-F1 ATP synthase making ATP, but instead they were used to extrude Na+ and Ca2+. So in a sense the mitochondrion can make ATP or extrude Ca2+. This becomes important when myocytes (or other cells) suffer from Ca2+ overload. In the short term, mitochondria can take up large amounts of Ca2+ to protect the cell from short-term Ca2+ overload, but chronic high [Ca2+]i has dire consequences. First, this Ca2+ uptake can diminish Ψm and occurs at the expense of ATP production (as noted), thus hampering energetic recovery from such stress. Second, elevated [Ca2+]i and [Ca2+]m can facilitate opening of the mitochondrial permeability transition pore, which allows the matrix contents to be released to the cytosol, wipes out Ψm, and can be the death knell for individual mitochondria, as well as the cells that rely on their function. Thus mitochondria can rapidly change into death-promoting organelles as just mentioned and also by producing excessive reactive oxygen species (ROSs), which can promote necrotic cell death via the mitochondrial permeability transition pore and release of proapoptotic proteins6 (see Chapter 22). Mitochondria can also induce mitochondrial autophagy, or mitophagy, which selectively clears damaged mitochondria and favors adaptation to stress by removing damaged mitochondria. Increased oxidative stress and apoptotic proteases can inactivate mitophagy and thereby cause cell death.7 The two chief contractile proteins are the motor protein myosin on the thick filament and actin on the thin filament (see Fig. 21-1). Ca2+ initiates the contraction cycle by binding to the thin filament regulatory protein troponin C to relieve the inhibition otherwise exerted by this troponin complex (Fig. 21-4). The thin actin filaments are connected to the Z-lines (Z, abbreviation for German Zuckung, or contraction; see Figs. 21-1 and 21-2) at either end of the sarcomere, which is the functional contractile unit that is repeated through the filaments. The sarcomere is limited on either side by a Z-line, which with the thin filaments creates a sort of cage around the thick myosin filament that extends from the center of the sarcomere outward toward, but not reaching the Z-line. During contraction, the myosin heads grab onto actin and pull the actin filaments toward the center of the sarcomere. The thin and thick filaments can thus slide over each other to shorten the sarcomere and cell length (without the individual actin or myosin molecules actually shortening). The interaction of the myosin heads with actin filaments when sufficient Ca2+ arrives from the SR (see Fig. 21-1) is called cross-bridge cycling. As the actin filaments move inward toward the center of the sarcomere, they draw the Z-lines closer together so that the sarcomere shortens. The energy for this shortening is provided by the breakdown of ATP, made chiefly in the mitochondria. Titin is a giant molecule, the largest protein yet described. It is an extraordinarily long, flexible, and slender myofibrillar protein (Fig. 21-5). Titin extends from the Z-line but stops just short of the M-line connecting the thick filament to the Z-line (see Fig. 21-1) and provides elasticity. Titin has two distinct segments: an inextensible anchoring segment and an extensible elastic segment that stretches as sarcomere length increases. So the titin molecule can stretch between 0.6 and 1.2 µm in length and has multiple functions. First, it tethers the myosin molecule to the Z-line, thereby stabilizing the contractile proteins. Second, as it stretches and relaxes, its elasticity contributes to the stress-strain relationship of cardiac and skeletal muscle. At short sarcomere lengths, the elastic domain is folded on itself to generate restoring force (Fig. 21-5). These changes in titin help explain the series elastic element that was inferred from mechanics studies as elasticity in series with the myosin filaments. Third, the increased diastolic stretch of titin as the length of the sarcomere in cardiac muscle is increased causes the enfolded part of the titin molecule to straighten. This stretched molecular spring then contracts more vigorously in systole.4 Fourth, titin may transduce mechanical stretch into growth signals. With sustained diastolic stretch, as in volume overload, the elastic segment of titin is under constant strain and transmits this mechanical signal to the muscle LIM protein (MLP) attached to the terminal part of titin that forms part of the Z-disc complex.8 MLP is proposed to be a stretch sensor that transmits the signals that result in the myocyte growth pattern characteristic of volume overload.9 This signal system may be defective in a subset of human dilated cardiomyopathy.9 Although at a molecular level the events underlying the cross-bridge cycle are complex, a prominent hypothesis holds that cross bridges exist in either a strong or a weak binding state (Fig. 21-6). The arrival of Ca2+ at the contractile proteins is a crucial link in excitation-contraction coupling. Binding of Ca2+ to troponin C shifts the troponin-tropomyosin complex on the actin filament, which permits the myosin heads to form strong binding cross bridges with actin molecules (Fig. 21-6). If, however, the strong binding state were continuously present, the contractile proteins could never relax. Thus it has been proposed that binding of ATP to the myosin head places the cross bridges in a weak binding state even when [Ca2+]i is high. Conversely, when ATP is hydrolyzed to ADP and inorganic phosphate (Pi), the strong binding state is again favored (Fig. 21-6). Hence the ATP-induced changes in the molecular configuration of the myosin head result in corresponding variations in its physical properties. Length-dependent activation also promotes the strong binding state (see the section Length-Dependent Activation and the Frank-Starling Effect). Conversely, the weak binding state predominates when [Ca2+]i falls and thereby allows relaxation during diastole. As Ca2+ dissociates from troponin C during the decline in [Ca2+]i, the troponin-tropomyosin complex resumes its inhibitory configuration to prevent strong binding. Although Ca2+ provides the essential “on” switch for the cross-bridge cycle by binding to troponin C, this is mediated by a series of interactions between components of the troponin complex, tropomyosin and actin (Fig. 21-6). To understand the role of Ca2+ requires a brief description of the molecular structure of actin and the troponin complex. The thin filaments are composed of two helical intertwining actin filaments, with a long tropomyosin molecule that spans seven actin monomers located in the groove between the two actin filaments (see Fig. 21-4). At every seven actin molecules (38.5 nm along this structure) sits a three-protein regulatory troponin complex: troponin C (Ca2+ binding), I (inhibitory), and T (tropomyosin binding). When [Ca2+]i is low, the tropomyosin molecule is positioned in such a way that it blocks the myosin heads from interacting with actin (see Fig. 21-4). As a result, most cross bridges are in the “blocked position,” although some might visit the weak binding state.4 As [Ca2+]i increases and binds with troponin C, troponin C binds more tightly to troponin I. This pulls the entire troponin complex (including troponin T) away from tropomyosin (Fig. 21-4), which allows tropomyosin to roll deeper into the thin filament groove,4 thereby largely disinhibiting the actin-myosin interaction. Thus weakly bound or blocked cross bridges enter the strongly bound state, and the cross-bridge cycle is initiated. As the strong cross bridges form, they nudge tropomyosin deeper into the actin groove. Tropomyosin contains evolutionarily conserved surface residues that are required for cooperative regulation of actomyosin.9 The troponin C–tropomyosin position at one site (open versus closed) also influences its “nearest-neighbor” sites and cooperatively spreads activation along the myofilament.2,4 Each myosin head is the terminal part of a heavy chain. The bodies of two of these chains intertwine and each terminates in a short “neck” that carries the elongated myosin head (see Fig. 21-4). According to the Rayment model, the base of the head, or the neck, changes configuration in the contractile cycle.8 Together with the “bodies” of all the other heads, the myosin thick filament is formed. Each lobe of the bilobed head has an ATP binding pocket (also called a nucleotide pocket) and a narrow cleft that extends from the base of this pocket to the actin-binding face.9 ATP and its breakdown products ADP and Pi bind to the nucleotide pocket in close proximity to the myosin site where ATP is split to ADP and Pi, (Fig. 21-6). The role of the narrow actin-binding cleft that splits the central 50-kDa segment of the myosin head in the contractile cycle is controversial. According to the revised Rayment model,4 this cleft responds to binding of ATP or its breakdown products to the nucleotide pocket in such a way that the conformational changes necessary for movement of the head are produced. According to Dominguez and colleagues,4 the cleft is closed in the weakly attached states before the power stroke (Fig. 21-6) but opens when Pi is released through the cleft, whereupon the myosin head attaches strongly to actin to induce the power stroke (Fig. 21-6D, E). Starting with the rigor state (Fig. 21-6A), binding of ATP to its pocket changes the molecular configuration of the myosin head such that the head detaches from actin to terminate the rigor state (Fig. 21-6B). Next, the ATPase activity of the myosin head splits ATP into ADP and Pi, and the head extends (Fig. 21-6C). As ATP is hydrolyzed, the myosin head binds to an adjacent actin unit. Pi is then released from the head through the cleft, and the myosin head is strongly bound to actin (Fig. 21-6D). Next, the head flexes through the power stroke, during which the actin molecule moves by approximately 10 nm,4 and the myosin head is now in the rigor state, which is sustained in the absence of ATP. When the pocket releases ADP and binds ATP, the cross bridge releases and the cycle repeats. During isometric (or isovolumic) contraction the cross bridges rotate but cannot fully move the actin filament, and the stretched strong-binding cross bridges bear force. During shortening (ejection) the actin filament moves during the power stroke. Each cycle of the cross bridge consumes one molecule of ATP, and this myosin ATPase activity is the major site of ATP consumption in the beating heart. Thus when the heart is more strongly activated (see later), the level of ATP consumption is similarly increased.4 Each myosin unit consists of two heavy chains with the bodies intertwined and each ending in one head. Notably, during contraction these two heads seem to work via a hand-over-hand action such that the myosin dimer never fully lets go of the thin filament during the activation period.10 There are two myosin isoforms in cardiac myocytes, alpha and beta, which have similar molecular weight but exhibit substantially different cross-bridge cycle and ATPase rates. The beta myosin heavy chain (β-MHC) isoform exhibits a slower ATPase rate and is the predominant form in adult humans. In small animals (rats and mice), the faster α-MHC form normally predominates but shifts to the β-MHC pattern during chronic stress and heart failure.4 Each myosin molecule neck also has two light chains (referred to as essential and regulatory light chains). The essential myosin light chain (MLC-1) is more proximal to the myosin head and may limit the contractile process by interaction with actin. The regulatory myosin light chain (MLC-2) is a potential site for phosphorylation, for example, in response to beta-adrenergic stimulation, and may promote cross-bridge cycling.11 In vascular smooth muscle, which lacks the troponin-tropomyosin complex, contraction is activated by the Ca2+-dependent myosin light chain kinase (MLCK) rather than by Ca2+ biding to troponin C (as in striated muscle). Myosin-binding protein C appears to traverse the myosin molecules in the A-band, thereby potentially tethering the myosin molecules and stabilizing the myosin head with respect to the thick and thin filaments. Defects in myosin, myosin-binding protein C, and several other myofilament proteins are genetically linked to familial hypertrophic cardiomyopathy.12 The myofilaments are activated in a graded rather than in all-or-none manner as a function of [Ca2+]i. The dynamics and regulation of Ca2+ transients in cardiac myocytes are discussed in the following section, but an important physiologic mechanism for regulating cardiac contractility (e.g., during sympathetic activity) is to increase peak [Ca2+]i and more fully activate the myofilaments. The higher the [Ca2+]i, the more fully saturated the Ca2+ binding sites on troponin C, and consequently more sites are available for cross bridges to form. When more cross bridges are working in parallel, the myocyte (and heart) can develop greater force. There is high cooperativity in this process, in large part because of the “near-neighbor” effect mentioned earlier. That is, Ca2+ bound to a single troponin C encourages local cross-bridge formation, and both Ca2+ binding and cross-bridge formation directly enhance the likelihood of cross-bridge formation in the 14 actin molecules controlled by one tropomyosin molecule. Furthermore, the openness of that domain directly enhances that of the neighboring domain with respect to both Ca2+ binding and cross-bridge formation. This cooperativity means that a small change in [Ca2+]i can have a large effect on the strength of contraction. Besides [Ca2+]i, the other major factor influencing the strength of contraction is sarcomere length at the end of diastole, just before the onset of systole. Both Otto Frank and Ernest Starling observed that the strength of the heartbeat was greater the more the diastolic filling of the heart. The increased heart volume translates into increased sarcomere length, which acts by a length-sensing mechanism.4 A part of this Frank-Starling effect has historically been ascribed to increasingly optimal overlap between the actin and myosin filaments. However, it has become clear that there is also a substantial increase in myofilament Ca2+ sensitivity with an increase in sarcomere length.4 A plausible mechanism for this regulatory change may reside in the decreasing interfilament spacing as the heart muscle is stretched.4 That is, the myocyte is at constant volume (over the cardiac cycle), so as the cell shortens, it must thicken, and conversely, when it is stretched, the cell gets thinner and filament spacing becomes narrower. This attractive lattice-dependent explanation for the Frank-Starling relationship has been challenged by the careful x-ray diffraction studies of de Tombe’s group.4 They found that reducing sarcomere lattice spacing by osmotic compression failed to influence myofilament Ca2+ sensitivity. Although several mechanisms could contribute to the Ca2+ sensitization of the myofilament at longer length, the issue is unresolved. When changes in diastolic length (or preload) are the cause of altered contractile strength, it is said to be a Frank-Starling (or sometimes just Starling) effect. Conditions in which contraction is strengthened (or weakened) independent of sarcomere length (e.g., increased Ca2+ transient amplitude) are referred to as positive (or negative) inotropic states or enhanced (or reduced) contractility. The distinction between these heterometric and homeometric mechanisms of altered cardiac strength is important. The cardiac cycle of Wiggers (see later) must be distinguished from the cross-bridge cycle.4 The former reflects the overall changes in pressure in the left ventricle, whereas the latter cycle is the repetitive interaction between myosin heads and actin. During isovolumic contraction (before aortic valve opening), the sarcomeres do not shorten appreciably, but cross bridges are developing force—not all simultaneously, however. That is, at any given moment some myosin heads will be flexing or flexed (resulting in force generation), some will be extending or extended, and some will be attached weakly to actin and some detached from actin. Numerous such cross-bridge cycles, each lasting microseconds, are integrated to produce the resulting force (and pressure). When ventricular pressure (sum of cross-bridge forces) reaches aortic pressure (afterload), ejection begins and is associated with the cross bridges actively moving the thin actin filaments toward the central area of the thick myosin filaments, thereby shortening the sarcomere. Note that as ejection proceeds (and sarcomeres shorten), myofilament Ca2+ sensitivity declines. Thus both [Ca2+]i decline and shortening cause a progressive decline in the contractile state as systole gives way to diastole. Both the Ca2+ transient properties and the myofilament Ca2+ sensitivity and cross-bridge cycling rate are altered under physiologic conditions (such as sympathetic stimulation and local acidosis or ischemia), which is discussed below and elsewhere. Volume and pressure overload may owe their different effects on myocardial growth to different patterns of force transmission.4 Whereas increased diastolic force is transmitted longitudinally via titin to reach the MLP protein, the postulated sensor (see the earlier section Titin and Length Sensing), increased systolic force may be transmitted laterally (i.e., at right angles) via the Z-disc and cytoplasmic actin to reach the cytoskeletal proteins and cell-to-matrix junctions such as the focal adhesion complex. How this mechanical force becomes translated into signals that activate the growth pathways such as those leading to mitogen-activated protein kinase (MAPK) and/or altered gene regulation and cell size and shape is addressed in other chapters. Genetic-based hypertrophic and dilated cardiomyopathies not only produce hearts that look and behave very differently but also have diverse molecular causes. These cardiomyopathies are in general linked to mutant genes that cause abnormalities in the force-generating system, such as β-MHC, troponin (T, I, and C), MLCs, myosin-binding protein C, and alpha-tropomyosin (see Chapter 65). One hypothesis is that mutations that increase contractile performance and/or energy demand result in concentric hypertrophy13 whereas mutations that either reduce force generation or result in non–force-generating cytoskeletal proteins (e.g., dystrophin, nuclear lamin, cytoplasmic actin, and titin) lead to a dilated cardiomyopathy. However, although the distinction between the two types of cardiomyopathy remains useful, it is oversimplified, with several examples of overlapping mechanisms. Ca2+ has a crucial role in regulating the contraction and relaxation phases of the cardiac cycle. Details of the associated Ca2+ fluxes that link contraction to the wave of excitation (excitation-contraction coupling) are now reasonably well clarified and accepted.2–4 Relatively small amounts of Ca2+ (trigger Ca2+) actually enter and leave the cardiomyocyte during each cardiac cycle, whereas larger amounts move in and out of the SR (Fig. 21-7). Each action potential depolarization traveling down the T tubules opens the voltage-gated L-type Ca2+ channels that are physically near the part of the SR lying close to the T tubule and activates SR Ca2+ release channels (RyRs). In this Ca2+-induced Ca2+ release mechanism, a smaller amount of Ca2+ entering via the Ca current (ICa) triggers the release of a relatively large amount of Ca2+ into the cytosol.2,4 In the human ventricle, SR Ca2+ release is three to four times higher than Ca2+ influx via ICa (in rat and mouse myocytes, amplification is two to three times higher).2 These Ca2+ fluxes elevate [Ca2+]i and promote binding of Ca2+ to troponin C and hence activation of the contractile process. Electron microscopic studies show that the SR is a continuous network surrounding the myofilaments with connections across Z-lines and transversely between myofibrils. There is direct functional evidence of continuity of the lumina of the entire SR network and nuclear envelope in adult cardiac myocytes. This allows relatively rapid diffusion of Ca2+ within the SR to balance free [Ca2+] within the SR ([Ca2+]SR).14 The total SR Ca2+ content is the sum of [Ca2+]SR plus substantially more bound to the intra-SR Ca2+ buffer calsequestrin. It is critical to both normal cardiac function and electrophysiology, and abnormalities contribute to systolic and diastolic dysfunction and arrhythmias. [Ca2+]SR dictates the SR Ca2+ content, the driving force for release of Ca2+, and regulates RyR release channel gating. The RyR channels that mediate release of Ca2+ from the SR are located at specialized junctions between the T tubule plasma membrane and the jSR membrane.2,4 Each junction has 50 to 250 RyR channels on the jSR that are directly under a cluster of 20 to 40 sarcolemmal L-type Ca2+ channels across a 15-nm junctional gap (that is actually crowded with protein). RyR2 (to denote the cardiac isoform) functions both as a Ca2+ channel and as a scaffolding protein that localizes numerous key regulatory proteins to the jSR.2,4 On the large cytosolic side, these include proteins that can stabilize RyR gating (e.g., calmodulin [CaM]; FK-506 binding protein [FKBP-12.6]); kinases that can regulate RyR gating by phosphorylation (e.g., protein kinase A [PKA] and Ca2+/CaM-dependent protein kinase II [CaMKII]); and the protein phosphatases PP1 and PP2A, which dephosphorylate the RyR (Fig. 21-8). Inside the SR the RyR also couples to several proteins (e.g., junctin, triadin, and via them calsequestrin) that similarly regulate RyR gating and, in the case of calsequestrin, provides a local reservoir of buffered Ca2+ close to the release channel. The actual RyR channel is made up of a symmetric tetramer of RyR molecules, each of which may have the aforementioned regulatory proteins associated with it. Thus the RyR receptor complex is very large (>7000 kDa; Fig. 21-8).15 When the T tubule is depolarized, one or more L-type Ca2+ channels open, and local cleft [Ca2+] increases sufficiently to activate at least one local jSR RyR (multiple channels here ensure high-fidelity signaling). The Ca2+ released from these first openings recruit additional RyRs in the junction via Ca2+-induced Ca2+ release to amplify release of Ca2+ into the junctional space. The Ca2+ diffuses out of that space throughout the sarcomere to activate contraction. Each of the approximately 20,000 jSR regions in the typical ventricular myocyte seems to function independently in response to local activation by ICa. Thus the global Ca2+ transient in the myocyte at each beat is the spatiotemporal summation of SR Ca2+ release events from thousands of jSR regions, synchronized by the upstroke of the action potential and activation of ICa. Ca2+-induced Ca2+ release is a positive feedback process, but it is now known that SR Ca release turns off when [Ca]SR drops by approximately 50% (i.e., from a diastolic value of ≈1 mM to a nadir of ≈400 µM).13 Elegant studies have documented how ICa is inactivated by high local [Ca2+], and this robust Ca-dependent inactivation is mediated by binding of Ca2+ to the CaM that is already associated with that channel. When Ca2+ binds to CaM, it alters channel conformation such that inactivation is favored. ICa is also subject to voltage-dependent inactivation during the action potential plateau, and thus inactivation limits further entry of Ca2+ into the cell. As for Ca2+-dependent RyR activation, several mechanisms may contribute to breaking its inherent positive feedback. First but not necessarily most compelling is analogous to Ca2+-CaM-dependent inactivation of ICa. That is, binding of Ca2+ to CaM that is prebound to RyR2 favors closure of the channel and inhibits reopening (Fig. 21-8).16 Second and undoubtedly important is that RyR2 gating is also sensitive to luminal [Ca2+]SR such that high [Ca2+]SR favors opening and low [Ca2+]SR favors closure.17 Indeed, release of Ca2+ from the SR during normal Ca2+ transients is robustly turned off when [Ca2+]SR falls to approximately half its normal value (≈400 µM, still 500 times higher than [Ca]i), almost regardless of the rate of SR Ca2+ release.13,14 A third and related factor is that as release proceeds and [Ca2+]SR declines, Ca2+ flux through the RyR falls and junctional [Ca2+] also falls, all of which tend to disrupt the positive feedback. That is, the RyR is less sensitive to activating Ca2+ (because [Ca2+]SR is low) and [Ca2+] on the activating side is also weaker.18 CaM has four Ca2+ binding sites, resembles troponin C, and participates in many different cellular pathways from ion channels to transcriptional regulation.16 In many cases (e.g., L-type Ca2+, Na+, and some K+ channels and RyR and inositol 1,4,5-triphosphate receptors), CaM is already prebound or “dedicated” such that elevation of local [Ca2+]i can rapidly induce Ca2+-CaM effects on their targets (Fig. 21-9).19,20 Indeed, more than 90% of the CaM in myocytes is already bound to cellular targets before Ca2+ binds to and activates it. Nevertheless, many myocyte CaM targets (e.g., CaMKII, calcineurin, nitric oxide synthase [NOS]) compete for this limited pool of “promiscuous” CaM. Thus CaM signaling in myocytes is complex and is further complicated by the effects of CaMKII, which influences some of the same targets and processes as CaM itself does.16,20 In addition to SR Ca2+ release triggered by ICa during normal excitation-contraction coupling, there is a finite probability that a given RyR will open stochastically. Because of local Ca2+-induced Ca2+ release in the junctional cleft, this can lead to spontaneous local SR Ca2+ release events known as Ca2+ sparks.18,21 Under normal resting conditions these Ca2+ sparks have a low probability (≈10−4), which means that at any moment there might be one or two Ca2+ sparks per myocyte. Because local [Ca2+]i declines rapidly as Ca2+ diffuses away from the initiating cleft, the resulting local [Ca2+]i at the next cleft (1 to 2 µm away) is normally too low to trigger that neighboring site. Thus Ca2+ sparks are very local events (within 2 µm in the cell). However, the probability of Ca2+ sparks is greatly enhanced when [Ca2+]i or [Ca2+]SR is elevated or under conditions in which the RyR is otherwise sensitized (e.g., by oxidation or CaMKII). These conditions can greatly enhance the likelihood that SR Ca2+ release from one junction will be sufficient to trigger neighboring junctions 1 to 2 µm away and result in propagating Ca2+ waves throughout the whole myocyte. These Ca2+ waves can be arrhythmogenic. First, the Ca2+ wave can activate substantial inward current via Na+/Ca2+ exchange (NCX; see later), which can depolarize the membrane potential and contribute to both early and delayed afterdepolarizations (EADs and DADs) during the action potential plateau or during diastole, respectively. EADs result in prolongation of the action potential duration, and DADs can initiate premature ventricular contractions (PVCs). Ca2+ is transported into the SR by SERCA, which constitutes almost 90% of the SR protein. Its molecular weight is approximately 115 kDa, and it straddles the SR membrane in such a way that part of it protrudes into the cytosol. Several isoforms exist, but in cardiac myocytes the dominant form is SERCA2a. For each molecule of ATP hydrolyzed by this enzyme, two calcium ions are taken up and accumulate within the SR (Fig. 21-10; also see Fig. 21-9). The source of the energy is at least in part derived from cytosolic generation of ATP via glycolysis.1 SR Ca2+ uptake is the primary driver of cardiac myocyte relaxation, and reuptake starts as soon as [Ca2+]i begins to rise. Because Ca2+ removal is slower than Ca2+ influx and release, a characteristic rise and fall in [Ca2+]i called the Ca2+ transient takes place. As [Ca2+]i falls, Ca2+ dissociates from troponin C and this progressively switches off the myofilaments. A reduction in SERCA expression or function (as seen in heart failure or energetic limitations) can thus directly result in slower rates of cardiac relaxation. In addition, the strength of SR Ca2+ uptake directly influences the diastolic SR Ca2+ content and [Ca2+]SR, which dictates both the sensitivity of the RyR and the flux rate of SR Ca2+ release. Thus SR Ca2+ uptake and release are an integrated system. Phospholamban (PLB) was so named by its discoverers Tada and Katz to mean “phosphate receiver.”23 PLB binds directly to SERCA2a, and under basal conditions this reduces the affinity of SERCA for cytosolic Ca2+, which results in weaker SR Ca2+ uptake at any given [Ca2+]i. However, when PLB is phosphorylated by either PKA or CaMKII (at Ser16 or Thr17, respectively), the inhibitory effect is relieved, thereby resulting in increased rates of SR Ca2+ uptake, cardiac relaxation (lusitropic effect), and increased SR Ca2+ content, which drives stronger contraction (inotropic effect; Fig. 21-10). The Ca2+ taken up into the SR is stored within the SR before further release. The highly charged low-affinity Ca2+ buffer (Kd ≈ 600 µM) calsequestrin is found primarily at the jSR and enhances the local availability of Ca for release via the nearby RyR. Calreticulin is another Ca2+-storing protein that is similar in structure to calsequestrin and probably similar in function. There is also evidence that calsequestrin and two other proteins located in the SR membrane (junctin and triadin) may regulate the properties of the RyR and be part of the mechanism by which higher [Ca]SR enhances RyR opening.17 Reuptake by SERCA occurs everywhere in the SR membrane in the network that surrounds the myofilaments. Diffusion of Ca2+ within the SR is relatively fast, which allows restoration of [Ca2+]SR at the jSR to occur quickly as Ca2+ is taken back up everywhere.24 Indeed, during normal Ca2+ release, intra-SR Ca2+ diffusion is rapid enough to limit Ca2+ gradients between SR release sites in the jSR and the Ca2+ uptake sites. This diffusion also ensures that [Ca2+]SR is relatively uniform throughout the myocyte, which facilitates the uniformity of SR Ca2+ release and myofilament activation throughout the cell. Excitation-contraction coupling is initiated by voltage-induced opening of the sarcolemmal L-type Ca2+
Mechanisms of Cardiac Contraction and Relaxation
Microanatomy of Contractile Cells and Proteins
Ultrastructure of Contractile Cells
Subcellular Microarchitecture
Mitochondrial Morphology and Function
Contractile Proteins
Titin and Length Sensing
Strong and Weak Binding States
Actin and Troponin Complex
Myosin and the Molecular Basis of Muscular Contraction
Graded Effects of [Ca2+]i on the Cross-Bridge Cycle
Length-Dependent Activation and the Frank-Starling Effect
Cross-Bridge Cycling Differs from the Cardiac Contraction-Relaxation Cycle
Force Transmission
Contractile Protein Defects and Cardiomyopathy
Calcium Ion Fluxes in the Cardiac Contraction-Relaxation Cycle
Calcium Movements and Excitation-Contraction Coupling
Ca2+ Release and Uptake by the Sarcoplasmic Reticulum
Sarcoplasmic Reticulum Network and Ca2+ Movements
Junctional Sarcoplasmic Reticulum and Ryanodine Receptor
Turning off Ca2+ Release: Breaking Positive Feedback
Calmodulin: A Versatile Mediator of Ca2+ Signaling
Calcium Sparks and Waves
Calcium Uptake into the Sarcoplasmic Reticulum by SERCA
Sarcolemmal Control of Ca2+ and Na+
Calcium and Sodium Channels
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Mechanisms of Cardiac Contraction and Relaxation
21
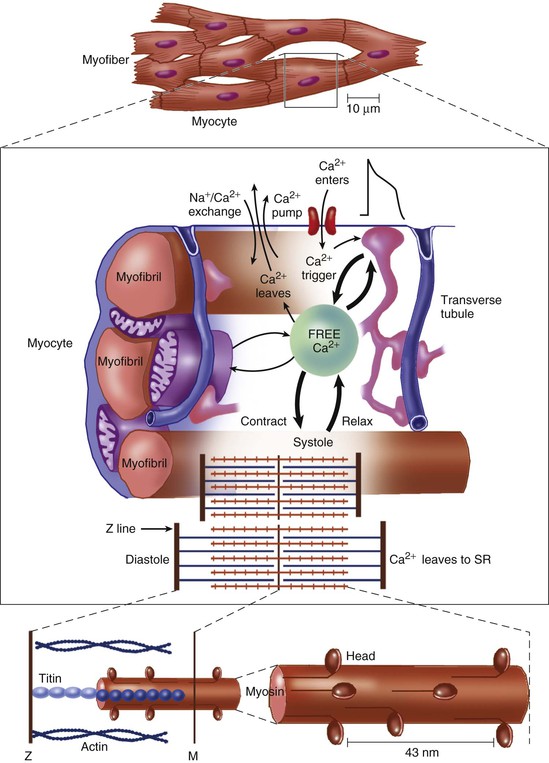
FIGURE 21-1 The crux of the contractile process lies in the changing [Ca2+] in the myocardial cytosol. Ca2+ ions are shown entering via the Ca2+ channel, which opens in response to the wave of depolarization that travels along the sarcolemma. These Ca2+ ions “trigger” the release of more Ca2+ from the SR and thereby initiate a contraction-relaxation cycle. Eventually, the small amount of Ca2+ that has entered the cell will leave predominantly by an Na+/Ca2+ exchanger, with a lesser role for the sarcolemmal Ca2+ pump. The varying actin-myosin overlap is shown for systole, when Ca2+ arrives, and for diastole, when Ca2+ leaves. The myosin heads, attached to the thick filaments, interact with the thin actin filaments, as shown in Figure 21-6. For role of titin, see Figure 21-5. The upper panel shows the difference between the myocardial cell or myocyte and the myofiber, which is composed of many myocytes. (Upper panel, Reproduced from Braunwald E, Ross J, Sonnenblick EH: Mechanisms of Contraction of the Normal and Failing Heart. 2nd ed. Boston, Little, Brown, 1976; other panels, reprinted from Opie LH: Heart Physiology, from Cell to Circulation. Philadelphia, Lippincott, Williams & Wilkins, 2004. Figure copyright L.H. Opie, © 2004.)
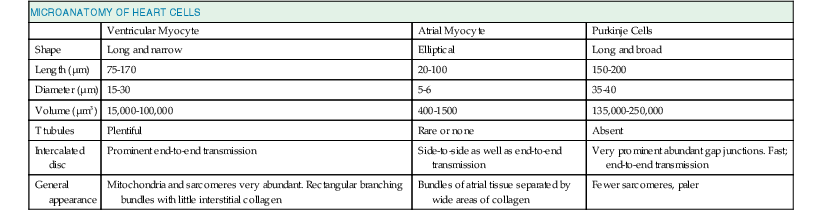
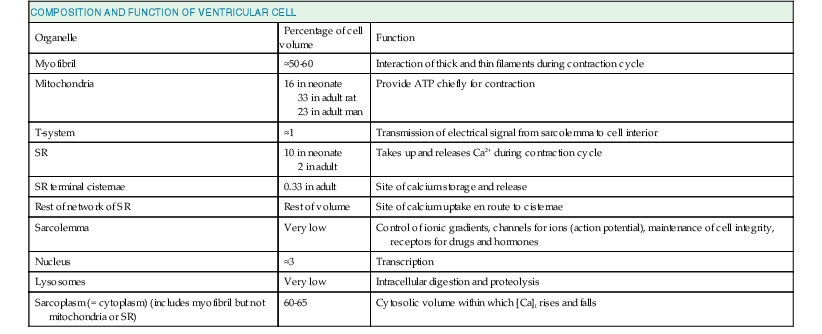
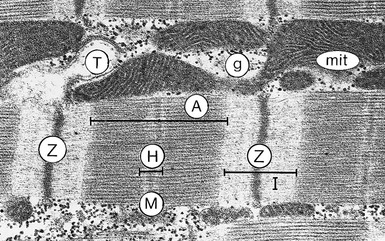
FIGURE 21-2 The sarcomere is the distance between the two Z-lines. Note the presence of numerous mitochondria (mit) sandwiched between the myofibrils and the presence of T tubules (T), which penetrate into the muscle at the level of the Z-lines. This two-dimensional picture should not disguise the fact that the Z-line is really a “Z-disc,” as is the M-line (M), also shown in Figure 21-1. A = band of actin-myosin overlap; g = glycogen granules; H = central clear zone containing only myosin filament bodies and the M-line; I = band of actin filaments, titin, and Z-line (rat papillary muscle, 32,000×). (Courtesy Dr. J. Moravec, Dijon, France.)
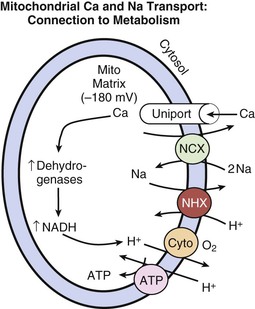
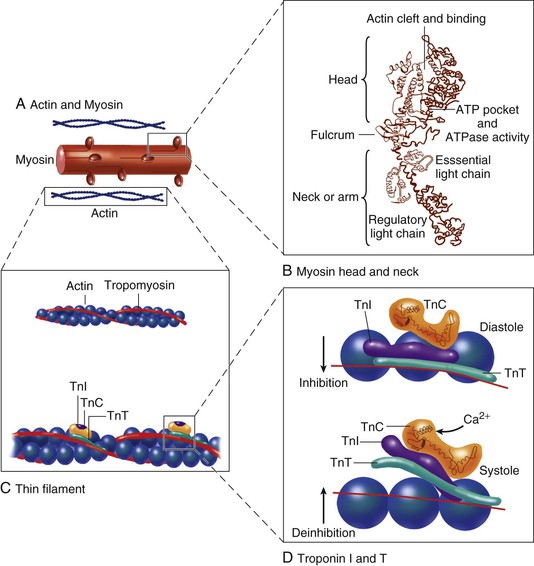
FIGURE 21-4 Major molecules of the contractile system. The thin actin filament (A) interacts with the myosin head (B) when Ca2+ ions arrive at troponin C (TnC) (C). A complex interaction between TnC and the other troponins moves tropomyosin to “uncover” an actin site to which a myosin head can attach. The molecular aspects are as follows. A, The thin actin filament contains TnC and its Ca2+ binding sites. When TnC is not activated by Ca2+, troponin I (TnI) inhibits the actin-myosin interaction. Troponin T (TnT) is an elongated protein that interacts with all the other components of the thin filament, thereby participating in the activation cycle (D). B, The molecular structure of the myosin head, based on Rayment and colleagues,8 is composed of heavy and light chains. The heavy head chain in turn has two major domains: one of 70 kDa (i.e., 70,000 molecular weight) that interacts with actin at the actin cleft and has an ATP binding pocket. The “neck” domain of 20 kDa, also called the “lever,” is an elongated alpha helix that extends and bends and has two light chains surrounding it as a collar. The essential light chain is part of the structure. The other regulatory light chain may respond to phosphorylation to influence the extent of the actin-myosin interaction. C, TnC with sites in the regulatory domain for activation by calcium and for interaction with TnI. D, Binding of calcium to TnC induces a conformational change in TnC, which elongates (compare systole with diastole). TnI closes up to TnC, and the normal inhibition of TnI on actin-tropomyosin is lessened; however, the interaction between TnC and TnT is strengthened. These changes allow repositioning of tropomyosin in relation to actin, with lessening of its normal inhibitory effects, as shown in the bottom panel. Now the contractile cycle can start. (Modified from Opie LH: Heart Physiology, from Cell to Circulation. Philadelphia, Lippincott, Williams & Wilkins, 2004. Figure copyright L. H. Opie, © 2004. D, Modified from Solaro RJ, Van Eyk J: Altered interactions among thin filament proteins modulate cardiac function. J Mol Cell Cardiol 28:217, 1999.)
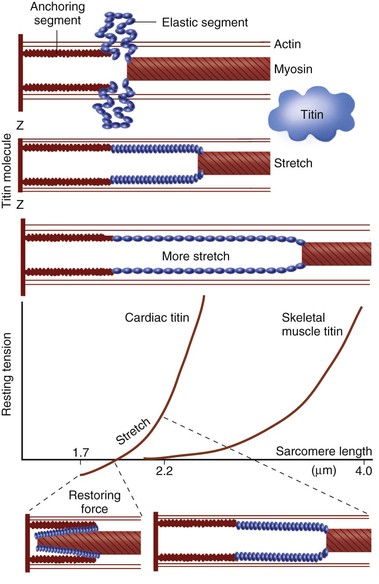
FIGURE 21-5 Titin, a very large elongated protein with elasticity, binds myosin to the Z-line. It may act as a bidirectional spring that develops passive force in stretched sarcomeres and resting force in shortened sarcomeres. As the sarcomere is stretched to its maximum physiologic diastolic length of 2.2 µm (see Fig. 21-18), titin first undergoes straightening (up to 2 µm) and then elongation, the latter rapidly increasing the passive force generated. At low sarcomere lengths, when sarcomeres are slack at approximately the diastolic limit of 1.85 µm (Fig. 21-18), the mechanically active elastic domain is folded on top of itself. At even shorter lengths, which may not be physiologic in an intact heart, substantial restoring force is generated. (Modified with permission of the American Heart Association from Trombitas K, Jian-Ping J, Granzier H: The mechanically active domain of titin in cardiac muscle. Circ Res 77:856, 1995; and Helmes M, Trombitas K, Granzier H: Titin develops restoring force in rat cardiac myocytes. Circ Res 79:619, 1996.)
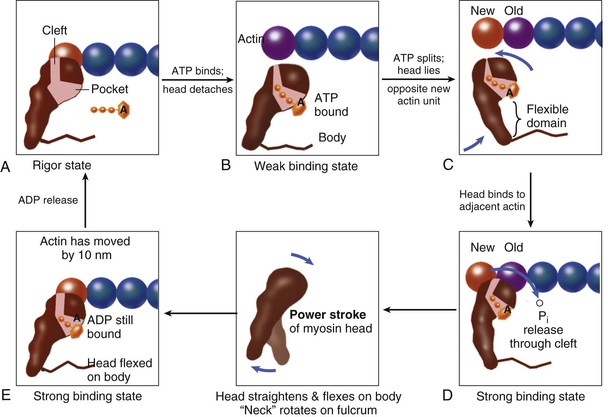
FIGURE 21-6 Cross-bridge cycling molecular model updated from the original Rayment five-step model for interaction between the myosin head and the actin filament8 that takes into account other models. The cross bridge (only one myosin head depicted) is pear shaped and consists of the catalytic motor domain, which interacts with the actin molecule, and an extended alpha helical “neck region,” which acts as a lever arm. The nucleotide pocket receiving and binding ATP is a depression near the center of the catalytic domain. The actin binding cleft bisects the catalytic motor domain. During the cross-bridge cycle, the width of the actin binding cleft changes in size, although details remain controversial. Starting with the rigor state (A), binding of ATP to the pocket (B) is followed by ATP hydrolysis (C), which partly closes the actin binding cleft. The cleft opens when phosphate is released (through the cleft rather than through the pocket), and the myosin head strongly attaches to actin to induce the power stroke (D, E). During the power stroke the latter rotates about a fulcrum in the region where the helix terminates within the catalytic motor domain. As the head flexes, the actin filament is displaced by approximately 10 nm (E). In the process ADP is also released, so the binding pocket becomes vacant. Finally, the rigor state is reached again (A) when the myosin head is once more ready to receive ATP to reinitiate the cross-bridge cycle. Throughout, the actin monomer with which the myosin head is interacting is speckled with dots. For references to Cooke, Holmes, and Dominguez, see Opie.4 Professor J.C. Rüegg of Heidelberg University, Germany, gave valuable advice.
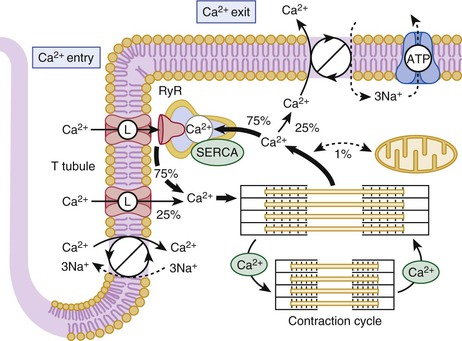
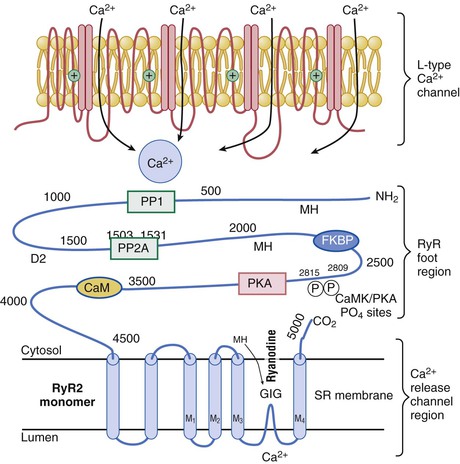
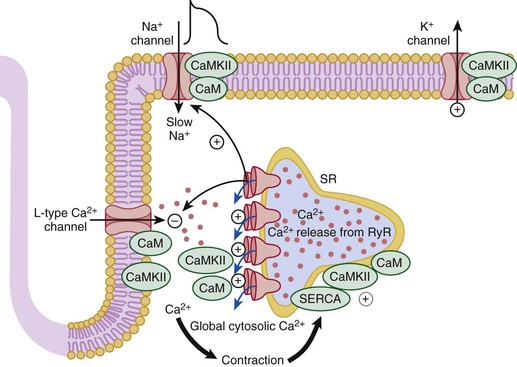
FIGURE 21-9 Role of CaM and its kinase in regulating intracellular [Ca2+]. The rising cytosolic Ca2+ concentration in systole activates the Ca2+ regulatory system whereby Ca2+-CaM causes inactivation of L-type Ca2+ current and RyR current. This negative feedback system limits cellular Ca2+ gain. The effects of CaMKII can also modulate these systems.19 For example, (1) CaMKII limits the extent of Ca2+-dependent inactivation and enhances Ca2+ current amplitude, (2) it increases the fraction of SR Ca2+ released from the RyR in response to the Ca2+ current trigger (which can be arrhythmogenic), (3) it phosphorylates PLB to enhance SR Ca2+ uptake by SERCA, and (4) it can modulate Na+ and K+ channel gating in ways that are also proarrhythmic.19,20
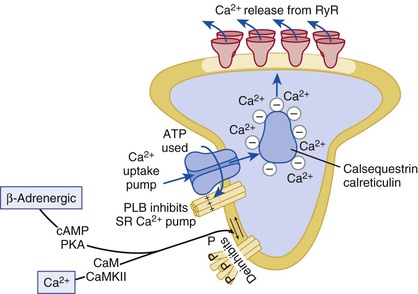
FIGURE 21-10 Ca2+ uptake into the SR by SERCA2a. An increased rate of uptake of Ca2+ into the SR enhances the rate of relaxation (lusitropic effect). PLB, when phosphorylated (P), removes the inhibition exerted on the Ca2+ pump by its dephosphorylated form. Thereby, Ca2+ uptake is increased either in response to enhanced cytosolic [Ca2+] or in response to beta-adrenergic agonists or CaMKII activation (which can be secondary to the beta-adrenergic system).2,20,22
