Chapter 8
Mechanisms of breathlessness
Dennis Jensen1,2, Kyle Pattinson3,4 and Caroline Jolley5
1Clinical Exercise and Respiratory Physiology Laboratory, Dept of Kinesiology and Physical Education, McGill University, Montreal, QC, Canada. 2Research Institute of the McGill University Health Centre, Translational Research in Respiratory Diseases Program, Montreal, QC, Canada. 3Nuffield Dept of Clinical Neurosciences, University of Oxford, John Radcliffe Hospital, Oxford, UK. 4Nuffield Dept of Anaesthetics, Oxford University Hospitals NHS Foundation Trust, John Radcliffe Hospital, Oxford, UK. 5Centre of Human and Aerospace Physiological Sciences, Faculty of Life Sciences and Medicine, King’s College London, London, UK.
Correspondence: Dennis Jensen, Dept of Kinesiology and Physical Education, McGill University, Montreal, QC, H2W 1S4, Canada. E-mail: dennis.jensen@mcgill.ca
The perception of breathlessness in the sensory (intensity) and affective (unpleasantness) domains involves the integration of respiratory efferent and afferent sensory information within cortical and subcortical neural networks. Recent advances in the visualisation of the neurobiology of breathing and physiological measurement of the neural respiratory drive (NRD) have furthered our understanding of breathlessness through the investigation of: 1) altered pulmonary physiology in symptomatic patients; and 2) the mechanisms of action of established and novel interventions. Irrespective of the underlying pathophysiology, the findings of such studies support the hypothesis that breathlessness intensity in obstructive and restrictive lung disorders reflects the awareness of increased NRD required to support ventilation, consequent to intrinsic respiratory mechanical loading and/or increased ventilatory demand. Qualitatively distinct descriptors of breathlessness (e.g. work/effort, air hunger, unsatisfied inspiration) probably involve the integration of discrete afferent inputs relative to the prevailing level of NRD. Conditioned responses are likely to underlie learned behaviours driving physical activity avoidance and breathlessness-related anticipatory fear and anxiety, contributing to the vicious cycle of functional decline in CRD.
Breathlessness is a prevalent and distressing symptom of pulmonary, cardiovascular, malignant and neuromuscular diseases [1]. Breathlessness has been defined as “a subjective experience of breathing discomfort that consists of qualitatively distinct sensations that vary in intensity. The experience derives from interactions among multiple physiological, psychological, social, and environmental factors, and may induce secondary physiological and behavioural responses.” [1] From this definition, it is clear that the mechanisms of breathlessness are highly complex and multifactorial. Using evidence from both experimental and clinical studies, this chapter focuses specifically on the central and peripheral physiological mechanisms of breathlessness and its key domains: the perceived intensity of breathlessness (sensory domain), unpleasantness (affective domain) and subsequent cognitive and emotional responses [2]. In particular, the chapter provides a review and discussion of: 1) the critical role of the neural respiratory drive (NRD) in causing and relieving breathlessness; 2) the role of central feedback from respiratory sensory afferent receptors in the neuromodulation of breathlessness and its clinical management; and 3) the central neuroanatomical structures underlying the perception of breathlessness in humans.
Neurophysiological mechanisms of breathlessness
NRD: the proximate cause of breathlessness
The output from the respiratory centres of the brainstem, the NRD, is tightly controlled to achieve the levels of minute ventilation (V′E) required to maintain blood gas homeostasis. Increases in the load imposed on the respiratory muscles and/or a reduction in their force-generating capacity, such as occurs in chronic obstructive and restrictive pulmonary disease, results in higher levels of NRD needed to support any given level of V′E at rest and during exercise (figure 1) [3–9]. Breathing is also under volitional cortical control, e.g. during talking and eating.
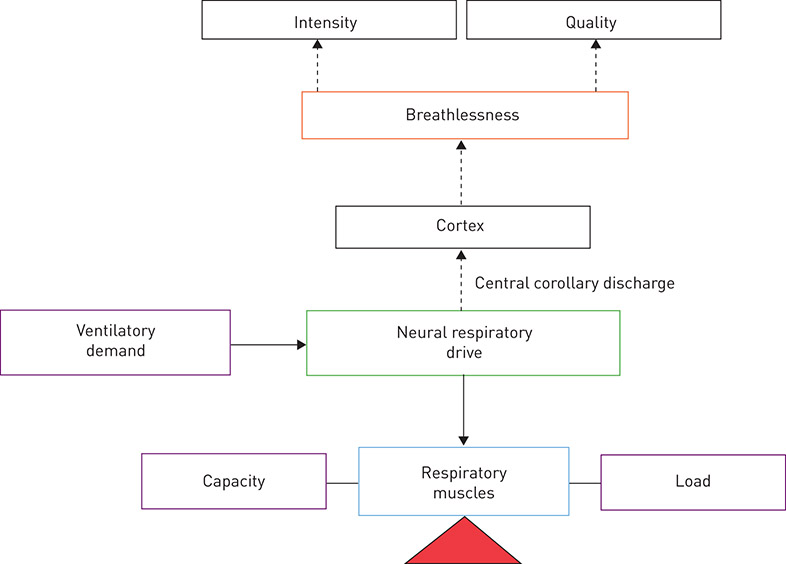
Figure 1. A physiological model of breathlessness in health and disease. Breathlessness, in both health and disease, can be rated quantitatively, in terms of intensity, and qualitatively in distinct sensory and affective domains (e.g. respiratory work/effort, air hunger, unsatisfied inspiration). Increased intrinsic mechanical loading, reduced capacity of the respiratory muscles and/or increased ventilatory demand leads to increased levels of neural respiratory drive to support ventilation. Increased neural respiratory drive results in increased breathlessness intensity. Thus, breathlessness intensity in cardiorespiratory disorders can be understood in the context of an appropriate integrated response to the combined effects of increased ventilatory demand and impaired ventilatory mechanics, which increase the mechanical load on the respiratory muscle pump and/or reduce respiratory muscle force-generating capacity. Adapted and modified from [9] with permission.
An increase in breathing implies not only an increase in NRD, but also generation of distinct patterns of sensory feedback from vagal afferents (innervating the lung and respiratory tract), and cervical and thoracic dorsal root afferents (innervating the proprioceptors of the chest wall and respiratory muscles) in response to thoracic volume expansion, respiratory muscle tension development and changes in respired flow and pressure (figure 2). Central feedback from respiratory sensory nerves contributes to the neuromodulation of breathlessness. For example, resumption of spontaneous breathing at the “break point” of a maximal voluntary breath-hold manoeuvre causes rapid relief of breathlessness (presumably via increased activity of vagal, cervical and/or thoracic dorsal root afferents), despite persistent or even increased chemostimulation (i.e. NRD) by hypoxia and hypercapnia [10, 11]. Moreover, an inverse relationship exists between tidal volume (VT) expansion and intensity ratings of air hunger at fixed elevated levels of end-tidal carbon dioxide tension (PETCO2) in mechanically ventilated, quadriplegic patients with high-level cervical injuries, an effect attributed to changes in pulmonary stretch receptor activity [12]. Afferent receptors of the pulmonary system most widely implicated in the neuromodulation of breathlessness include: central and peripheral chemoreceptors; slowly and rapidly adapting stretch receptors; bronchopulmonary C-fibres, including juxtacapillary or J-receptors; and chest wall and respiratory muscle spindles and Golgi tendon organs [13–16]. A detailed description of the morphological and functional features of these afferent receptors and their effective stimuli have been reviewed in detail elsewhere [16, 17].
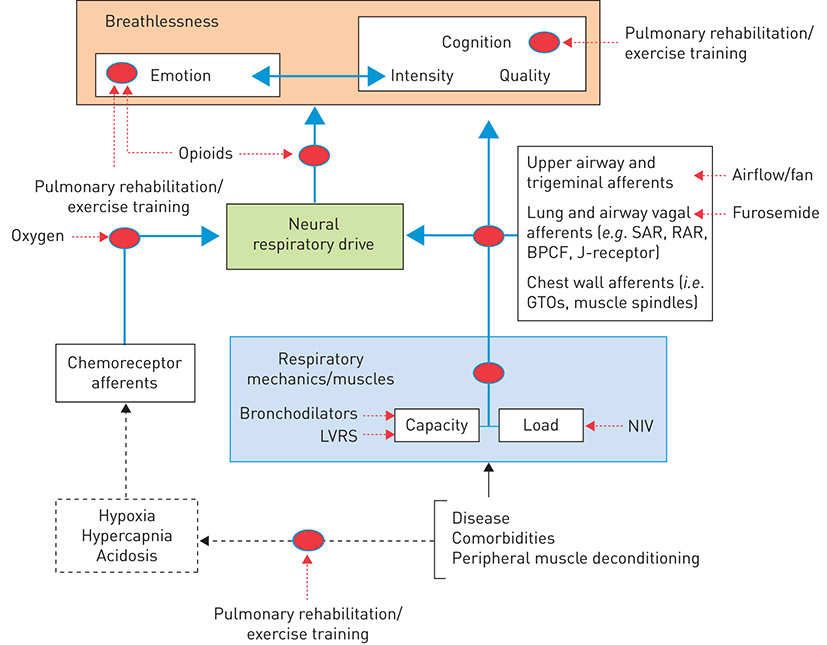
Figure 2. A simplified conceptual neurophysiological model of breathlessness in the sensory (intensity), affective (unpleasantness) and emotional domains, with selected pharmacological and non-pharmacological targets and their proposed mechanism(s) of action. As illustrated in figure 1, increased intrinsic respiratory mechanical loading, reduced respiratory muscle force-generating capacity and/or increased ventilatory demand result in increased neural respiratory drive, which is perceived as increased breathlessness intensity. The corollary of this is that any intervention capable of reducing neural respiratory drive by improving the load–capacity balance of the respiratory muscle pump (e.g. bronchodilators, LVRS, NIV) and/or decreasing ventilatory demand (e.g. oxygen, rehabilitative exercise training) has the potential to alleviate breathlessness intensity in patients with respiratory disease. Refer to the text for further details. LVRS: lung volume reduction surgery; SAR: slowly adapting stretch receptor; RAR: rapidly adapting stretch receptor; BPCF: bronchopulmonary C-fibre; J-receptor: juxtacapillary receptor; GTO: Golgi tendon organ.
Transmission of sensory afferent information to higher cortical centres provides a potential mechanism by which afferent feedback can modulate breathlessness perception directly [18, 19]. However, the homeostatic function of sensory feedback from afferent receptors, in concert with central and peripheral chemoreceptor inputs, is to modulate efferent motor output to the respiratory muscles, i.e. NRD [19]. “Central corollary discharge” refers to the simultaneous projection of resultant neural signals from the motor cortex and/or respiratory centres of the brainstem to the respiratory pump muscles and sensory areas of the brain. Studies by CHEN et al. [20, 21] in unanaesthetised, decerebrated, vagus- and carotid sinus-denervated, paralysed and ventilated cats reported that thalamic and mesencephalic neurons demonstrate phasic respiratory activity, which increased as a function of increasing phrenic nerve discharge and thus NRD. These findings were interpreted as evidence of respiratory-related central corollary discharge and led to the hypothesis that the perception of breathlessness in humans is mechanistically linked to the awareness of increased NRD as sensed by increased central corollary discharge. Support for this hypothesis comes from studies in healthy adults wherein breathlessness intensity ratings increased swiftly to intolerable levels during voluntary breath holding (which is associated with progressive increases in NRD due to central and peripheral chemoreflex stimulation by asphyxia), despite little or no activation of respiratory sensory afferents due to the cessation of breathing [10, 22]. Moreover, the intensity of perceived air hunger, an inherently unpleasant form of breathlessness [23], increases in direct proportion to elevations in PETCO2 (reflecting increased chemostimulation and thus NRD) at fixed levels of V′E in mechanically ventilated quadriplegic patients with complete paralysis resulting from high-level cervical injuries [24] or neuromuscular blockade [23, 25]. Collectively, these findings suggest that: 1) increased NRD is the proximate cause of breathlessness; and 2) simultaneous feedback from central and peripheral chemoreceptors, and vagal, cervical and thoracic dorsal root afferent nerves contribute to the neuromodulation of breathlessness via their effects on NRD.
In this context, the intensity of perceived breathlessness in disease states can be understood as the awareness of the increased NRD needed to support a given level of V′E in the setting of increased respiratory mechanical loading and/or reduced force-generating capacity of the respiratory muscles (figure 1). Qualitatively distinct descriptors (e.g. chest tightness, work/effort) and unpleasant affective dimensions of breathlessness (e.g. air hunger, unsatisfied inspiration), which can be rated independently of the sensory intensity domains of breathlessness, are considered to result from the integration of distinct afferent sources [1, 2, 23, 26–28]. For example, DEMEDIUK et al. [27] found that increasing NRD by increasing PETCO2 at a constant V′E decreased the intensity ratings of respiratory work/effort and simultaneously increased the intensity ratings of air hunger. LANSING et al. [28] similarly reported that the intensity ratings of air hunger (not respiratory work/effort) increased when NRD was increased via increased PETCO2 at a constant V′E, whereas the intensity ratings of respiratory work/effort (not air hunger) increased when V′E was increased at a constant PETCO2. In keeping with these observations, BANZETT et al. [23] showed that air hunger is far more unpleasant than respiratory work/effort at matched levels of intensity.
Central neuroanatomical substrates of breathlessness
As outlined earlier, the conscious perception of breathlessness reflects a complex integration of efferent and afferent sensory information shaped by conscious and unconscious processes in the brain (figure 2). Visualisation of the neurobiology of breathlessness through functional neuroimaging is a relatively new field of research [29, 30]. The brain area most commonly identified during laboratory-induced breathlessness is the insular cortex (often extending into the operculum) [31–42]. The insular cortex is associated with emotion processing, self-awareness and consciousness [43, 44], and is part of a network that integrates sensory information with conscious and unconscious processes such as decision-making, emotions, suffering, memory, motivation and arousal. The prefrontal cortex, anterior and posterior cingulate cortices, cerebellum, amygdala, striatum and periaqueductal grey matter (PAG) have all been inconsistently identified in the various neuroimaging studies of breathlessness [18, 34, 37–42, 45–52].
The interpretation and generalisability of findings of functional neuroimaging studies of breathlessness is limited because of the varying methodologies used and the relatively small number (i.e. <20) of original research articles. Furthermore, the extent to which the findings of studies investigating the mechanisms of acute laboratory-induced breathlessness in healthy adults [18, 49] are applicable to understanding the neurobiology of chronic breathlessness in advanced disease is unclear; however, studies in symptomatic patients with COPD are starting to emerge (figure 3) [48]. The discussion that follows proposes how the aforementioned areas of the brain may contribute to breathlessness, although in most cases definitive neuroimaging studies are required.
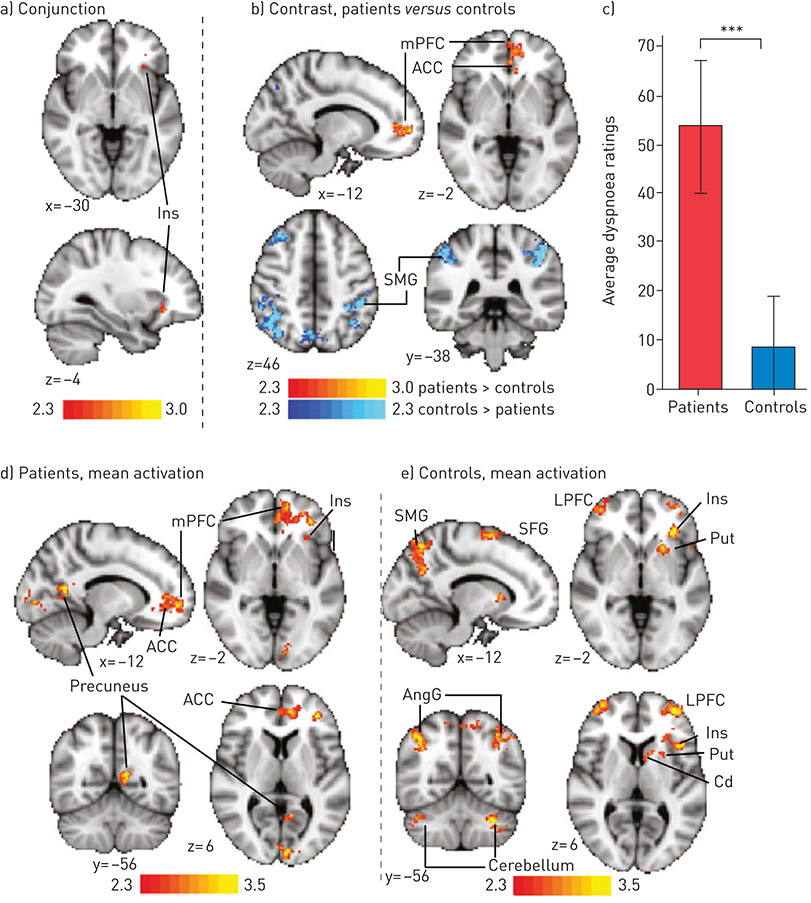
Figure 3. Activation (contrasts and conjunction) correlating with VAS ratings to breathlessness-related word cues in people with COPD and age-sex matched healthy controls. This demonstrates activation of many emotion-regulating areas. Maps are cluster-level corrected for multiple comparisons at p<0.05 across the whole brain. Maps represent conjunction analysis (activations common to both groups), comparisons between groups (patients versus controls in red-yellow, controls versus patients in blue-light blue), and mean activations in patients and controls. Bar graph is mean±SD breathlessness ratings for each group. ***: p<0.001. Cerebellum is crus I and VI. ACC: anterior cingulate cortex; AngG: angular gyrus; Cd: caudate; Ins: insular cortex; LPFC: lateral prefrontal cortex; MFG: middle frontal gyrus; mPFC: medial prefrontal cortex; Put: putamen; SMG: supramarginal gyrus; SFG: superior frontal gyrus. Reproduced from [48] with permission.
By driving breathlessness-related fear and anxiety, maladaptive learning may be a key mechanism underlying the severity of chronic breathlessness, regardless of disease status [53]. Learned associations between otherwise innocuous cues (e.g. the ring of a doorbell) and breathlessness during the behavioural response (e.g. walking to answer the door) exemplify the psychological process known as conditioning. Conditioned responses are likely to underlie learned behaviours driving physical activity avoidance and breathlessness-related anticipatory fear and anxiety, contributing to the well-described vicious cycle of functional decline in CRD. Drawing on observations in the pain literature [54, 55], the insular, prefrontal and anterior cingulate cortices may possibly work together to prepare the brain for an impending breathlessness stimulus. To this end, a recent report by FAULL et al. [56] provided new evidence for a role of the PAG, which gates neural traffic to and from the periphery (e.g. vagal afferent receptors), in conditioned breathlessness anticipation in healthy adults.
Depression is known to worsen pain via actions in the prefrontal cortex [57] as part of a “suffering” network that interacts with the insular cortex [58]. Depression is also associated with heightened symptoms of breathlessness [59]. In a recent study of brain responses to breathlessness-related word cues in patients with COPD [48], activity in the prefrontal and paracingulate cortices was correlated with ratings of breathlessness (figure 3) and partly explained by depression levels. Another study by VON LEUPOLDT et al. [39] used a focused region of interest approach to investigate the impact of mood on activity in the amygdala and insular cortex of healthy volunteers. In that study, mood was manipulated by presenting happy and sad faces during resistive load breathing, with stronger breathlessness-related activation of the amygdala and insular cortex occurring during sad versus happy face presentation [39]. It is worth considering, however, that happy or sad faces might activate the amygdala and insular cortex, in a manner independent of the breathlessness stimulus.
One new technology that might help advance our understanding of breathlessness processing is magnetoencephalography (MEG), which measures brain activity by detecting magnetic fields generated by neuronal currents [60, 61]. The particular advantages of MEG for the study of breathlessness are that it has a high temporal resolution and experiments are conducted in the sitting position. JOHNSON et al. [61] recently confirmed that MEG is a feasible method to investigate exercise-induced breathlessness in adults with chronic lung disease. Future work using MEG would be particularly suited to investigating the dynamics of brain networks that process breathlessness and its response to therapy.
Understanding breathlessness through pulmonary disease pathophysiology
Obstructive lung disease
In diseases characterised by expiratory airflow limitation (e.g. COPD, asthma, cystic fibrosis (CF)), NRD is elevated at rest and during exercise compared to healthy individuals to compensate for the increased respiratory mechanical load relative to reduced respiratory muscle force-generating capacity, these being consequences of impaired respiratory mechanics (figures 4 and 5a) [3–9, 62]. Ventilatory demand is also increased to overcome ventilation–perfusion mismatch [63], hypoxaemia, hypercapnia and early-onset metabolic (lactic) acidosis during exercise as a consequence of the reduced aerobic capacity of the locomotor muscle [64, 65].
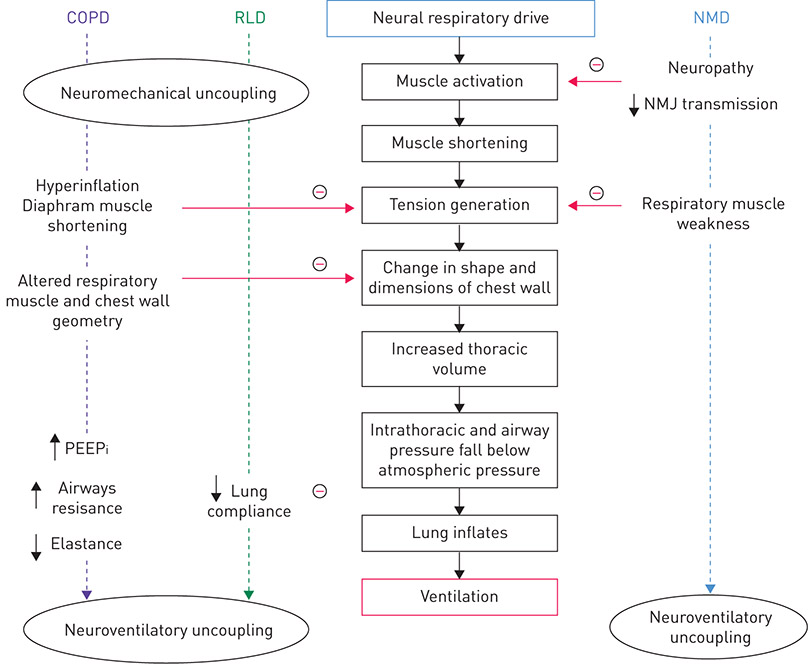
Figure 4. Impact of COPD, restrictive lung disease (RLD, e.g. ILD) and neuromuscular disease complicated by respiratory muscle weakness (e.g. amyotrophic lateral sclerosis (ALS)) on the translation of neural respiratory drive to ventilation. Disordered ventilatory mechanics in patients with COPD and RLD progressively uncouple the neural respiratory drive from generation of respiratory muscle tension, intrathoracic pressure and ventilation, i.e. neuromechanical and neuroventilatory uncoupling. In neuromuscular diseases, respiratory muscle weakness causes neuroventilatory uncoupling as a result of neuropathy (e.g. ALS), impaired neuromuscular junction (NMJ) transmission (e.g. myasthenia gravis), a primary muscle disorder (e.g. Duchenne’s muscular dystrophy) or a mixed neuropathic/myopathic disorder (e.g. critical illness neuromyopathy). PEEPi: intrinsic positive end-expiratory pressure; NMD: neuromuscular disease. Adapted and modified from [9] with permission.
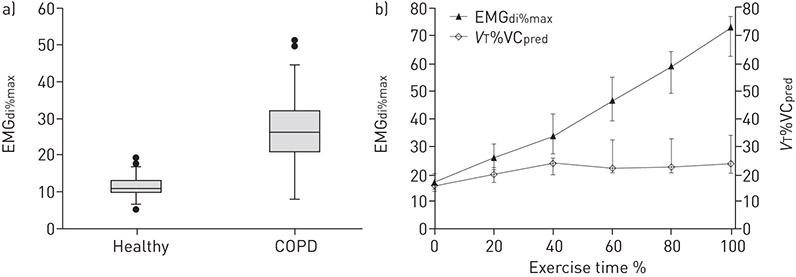
Figure 5. a) Box-and-whisker plots comparing resting measures of neural respiratory drive (i.e. diaphragm electromyogram activity expressed as a percentage of volitional maximum (EMGdi%max)) in 30 patients with COPD, with 26 healthy individuals matched for age, height and BMI (p<0.001 by independent samples t-test). The box length is the interquartile range (IQR). Closed circles are outliers, i.e. cases with values between 1.5 and 3 IQRs from the upper and lower edge of the box. Reproduced from [3] with permission. b) Relationship between increasing neural respiratory drive (EMGdi%max) and increasing thoracic volume displacement (tidal volume (VT) expressed as a percentage of predicted vital capacity (VCpred) (VT%VCpred)) during symptom-limited incremental cycle or treadmill exercise testing in 12 patients with COPD. Data points represent median and IQR. Reproduced from [72] with permission.
The capacity of the respiratory system to respond to the increased ventilatory demands of exercise is grossly impaired in adults with COPD, particularly those with advanced disease [65]. This impairment is due to static and dynamic lung hyperinflation (figure 6) [66], which positions VT on the upper alinear (noncompliant) extreme of the respiratory system’s sigmoid pressure–volume relationship. Under these circumstances, VT expansion becomes mechanically constrained early in exercise as inspiratory capacity (IC) and inspiratory reserve volume (IRV) decrease from their relatively reduced resting values (figure 7c–e) [66]; breathing frequency increases (tachypnoea) (figure 7d) [66]; the inspiratory muscles (e.g. diaphragm) shorten, functionally weaken and become less contractile [67–70]; respiratory muscle effort requirements and the work of breathing increase [62, 71]; and breathlessness intensity ratings rapidly escalate to intolerable levels (figure 8) [66].
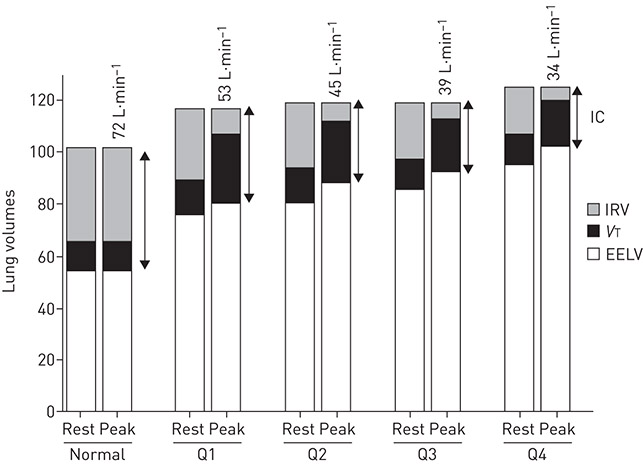
Figure 6. Progressive hyperinflation, shown by increasing end-expiratory lung volume (EELV), expressed as % predicted total lung capacity, is illustrated at rest and peak exercise as FEV1 quartile worsens in patients with COPD. Peak values of dynamic inspiratory capacity (IC), tidal volume (VT) and ventilation (values shown above peak exercise bars) decreased with worsening disease severity, although similar peak ratings of breathlessness intensity were reached (data not shown). Normative data are shown for comparison (n=21). Data across groups of patients with COPD (n=427) were divided into quartiles based on FEV1 expressed as a percentage of predicted normal values, where Q1=54.5–85.1%; Q2=43.8–54.1%; Q3=34.9–43.6%; and Q4=16.5–34.9%. IRV: inspiratory reserve volume. Reproduced from [66] with permission.
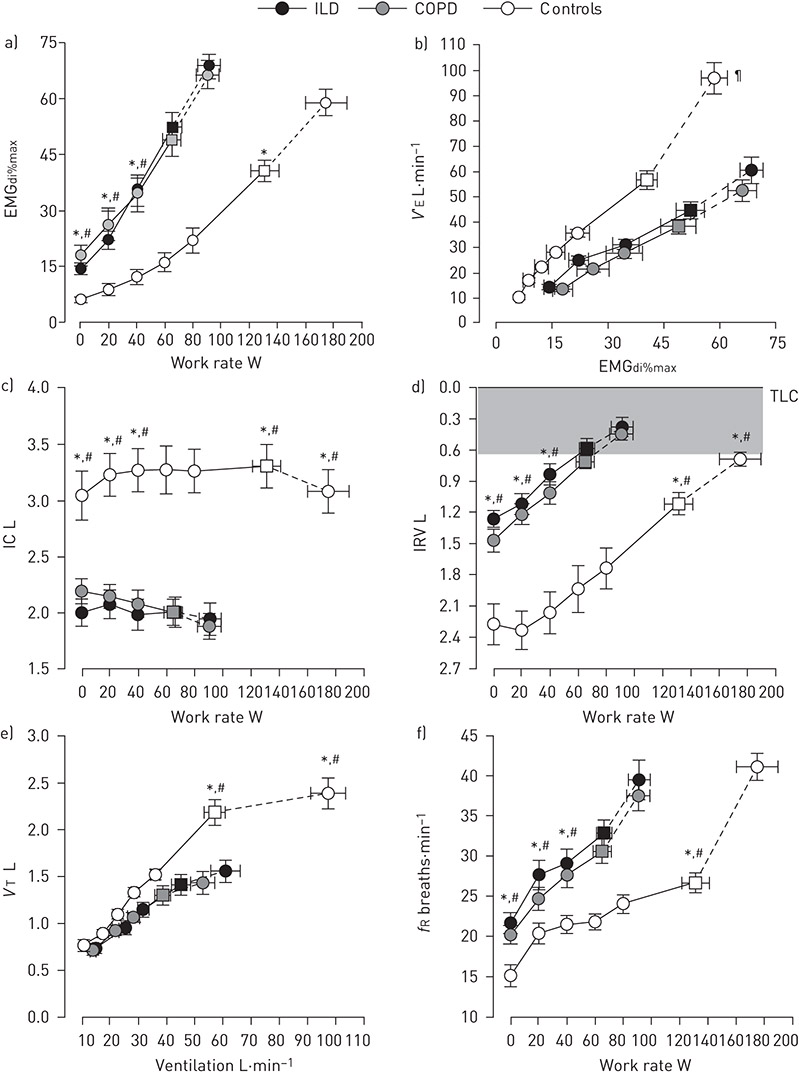
Figure 7. a) Neural respiratory drive, b) neuroventilatory coupling, c) and d) operating lung volumes, and e) and f) breathing pattern during symptom-limited incremental cycle exercise testing in patients with ILD (n=16), patients with COPD (n=16), and age-matched healthy controls (n=16). The shaded area in d) represents the minimal inspiratory reserve volume (IRV) reached by all groups at the end of exercise. Square symbols represent tidal volume (VT)-minute ventilation (V′E) inflection points. Values are mean±SEM. *: p<0.05 for ILD versus controls; #: p<0.05 for COPD versus controls; ¶: p<0.05 for differences in V′E-neural respiratory drive slopes between patient groups and healthy controls. EMGdi%max: root mean square of the crural diaphragm electromyogram activity expressed as a percentage of volitional maximum; IC: inspiratory capacity; fR: breathing frequency; TLC: total lung capacity. Adapted from [5] with permission.
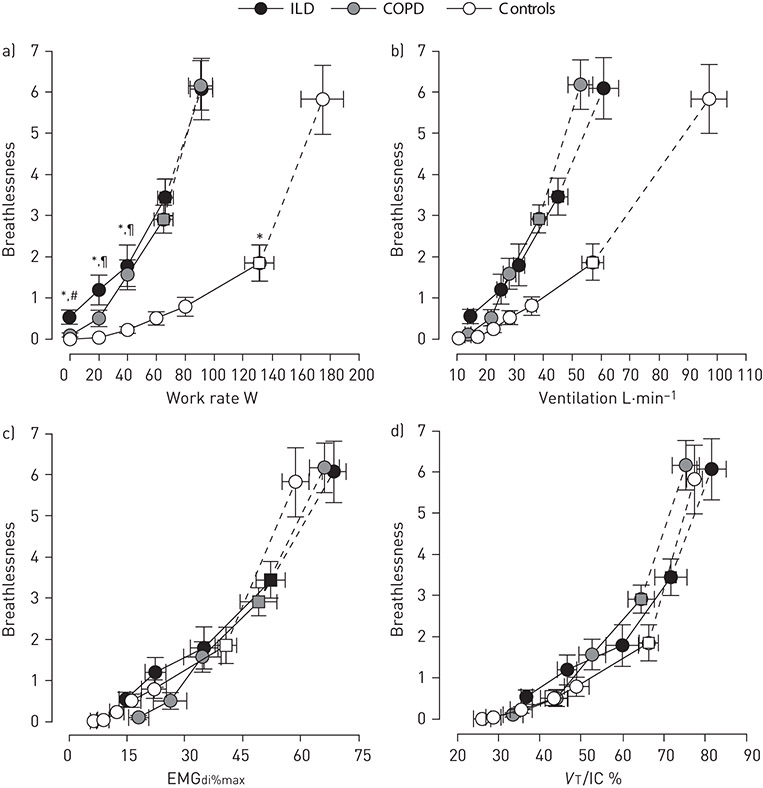
Figure 8. Exertional breathlessness (breathlessness) intensity ratings during symptom-limited incremental cycle exercise testing in patients with ILD (n=16), patients with COPD (n=16), and age-matched healthy controls (n=16). Breathlessness intensity ratings (Borg scale) are presented relative to: a) work rate; b) ventilation; c) neural respiratory drive; and d) tidal volume (VT) to inspiratory capacity (IC) ratio. Square symbols represent the VT-ventilation inflection points. Values are mean±SEM. *: p<0.05 for ILD versus controls; #: p<0.05 for ILD versus COPD; ¶: p<0.05 for COPD versus controls. EMGdi%max: root mean square of the crural diaphragm electromyogram activity expressed as a percentage of volitional maximum. Reproduced from [5] with permission.
The progressive shortening, weakening and mechanical loading of the inspiratory muscles that accompany exercise-induced decreases in IC and IRV necessitate, in the face of increased ventilatory demand, a compensatory increase in NRD to generate the intrathoracic pressures and respiratory flow rates needed to achieve or maintain any given VT and V′E during exercise in patients with COPD versus healthy individuals (figures 4, 7a and 7b) [5, 6, 62, 67, 72]. The resultant disparity between increasing NRD and increasing mechanical/muscular output of the respiratory system (figures 5b and 7b) has been termed neuromechanical/neuroventilatory uncoupling or dissociation.
A growing body of evidence suggests that higher breathlessness intensity ratings at equivalent power outputs and levels of V′E during exercise in patients with CF and COPD (figure 8a and 8b) versus healthy controls are mechanistically linked to higher NRD, quantified as diaphragm electromyogram (EMGdi) activity expressed as a percentage of volitional maximum (EMGdi%max) (figure 8c) [4, 5, 62, 72]. MURPHY et al. [73] similarly reported that changes in NRD, quantified as parasternal intercostal EMG activity expressed as a percentage of volitional maximum, following an acute exacerbation of COPD correlated positively with concurrent changes in breathlessness intensity ratings, further substantiating the role of increased NRD as the proximate cause of breathlessness in patients with obstructive lung disease.
ILD
Pulmonary reserve in ILD is limited by mechanical and gas exchange abnormalities. This results in increased NRD as a consequence of increased ventilatory demand and increased mechanical load on the respiratory muscles, increasing the work of breathing [5, 74–76]. Respiratory muscle weakness should also be considered as a cause of reduced ventilatory capacity in ILD [74, 77–79], particularly in the context of inflammatory myopathies [80].
In persons with ILD, the sigmoid pressure–volume curve of the respiratory system typically shifts downwards and to the right (i.e. contracts along its volume axis), which is consistent with a stiff, noncompliant lung (figure 4) [81]. As a result, pulmonary function tests in ILD are consistent with a restrictive ventilatory defect (i.e. reduced total lung capacity, vital capacity, IC and IRV) without reductions in isovolume flow rates [5, 77, 81, 82]. Lung gas transfer is frequently reduced and its decline may precede abnormalities in lung volume [83]. The main cause for exercise-induced hypoxaemia is ventilation–perfusion mismatch [84].
Compared with healthy controls, patients with ILD report higher levels of breathlessness intensity at any given V′E, power output and rate of oxygen uptake (V′O2) during exercise (figure 8a and 8b) [5, 82]. In addition to increasing the intrinsic mechanical loading of the respiratory muscles, these impairments in static and dynamic ventilatory mechanics (e.g. reduced IC and IRV) also serve to limit VT expansion early in exercise despite increasing NRD (figure 7c–e) [5, 82]. The inability to further expand VT means that increases in V′E can only be achieved through increases in breathing frequency (tachypnoea) (figure 7f). Using EMGdi%max to quantify NRD, FAISAL et al. [5
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


