Figure 7–1.
(a) Phases of a cardiac action potential and ion channel currents. (b) blocking effects of Lidocaine and Quinidine on action potential duration
A number of classification systems of antiarrhythmic drugs based on their interaction with ion channels and receptors have been proposed including that by Vaughan-Williams [1]. Vaughan-Williams first proposed a scheme based on electrophysiological mechanisms of drug action to classify antiarrhythmic drug action [2]. Although knowledge about the electrophysiological bases of arrhythmias and drug action at that time were limited, this classification is still commonly used. Vaughan-Williams classification divides antiarrhythmic drugs into four major groups (Class I–IV) according to whether their major effect is to block Na+ channel (Class I drug), β-adrenergic receptor (Class II drug), K+ channel (Class III drug) or Ca2+ channel (Class IV drug) (Table 7.1). In general, by blocking the Na+ channel, Class I drugs reduce maximum rate of rise in phase 0 of the AP without changing the resting potential, but have different effects on action potential duration (APD). The Class I drugs are further divided into three subgroups (Class Ia, Ib, Ic) according to their different effects on APD: Class Ia drugs prolong APD, Class Ib drugs shorten APD, and Class Ic drugs do not have significant effects on APD (Fig. 7.1b). Class II drugs block β-adrenergic receptors. Class III drugs block K+ channels of delayed-rectifier type and prolong APD. Class IV drugs block Ca2+ channels. Vaughan-Williams classification has the virtue of simplicity. However, many anti-arrhythmic drugs may block more than one type of ion-channel. This classification cannot account for such complex phenomena and, moreover some antiarrhythmic agents including digitalis and adenosine cannot be covered by the four groups. One important classification of many others proposed to deal with these problems is the Sicilian Gambit [3]. The working group of the European Society of Cardiology met in Taormina, Sicily to consider the classification of anti-arrhythmic drugs [3]. They criticized the Vaughan-Williams classification because of the following reasons: (1) The classification is a hybrid. A single class effect can be produced by multiple mechanisms and some drugs have several classes of actions. (2) Activation of channels or receptors is not considered. (3) The classification is incomplete. For example, α-adrenergic blockers, cholinergic agonists, digitalis and adenosine are not included. The Sicilian Gambit group proposed that the Vaughan-Williams classification system be replaced with a new classification. In the new classification, the vulnerable parameters associated with specific arrhythmic mechanisms are identified and the effects of each drug on each parameter are listed to characterize the profile of each drug in the termination or suppression of the arrhythmia depending on its underlying mechanisms (Table 7.2). Thus, a most effective drug with possible minimal possible adverse effects would be expected. This system can account for multiple drug actions and provides more flexibility for classifying anti-arrhythmic drugs. However, this multidimensional classification system is significantly more complex than the standard Vaughan-Williams classification and may not be suitable for a basic understanding of antiarrhythmic drug actions. Because of its simplicity, the Vaughan-Williams classification will be used in this chapter to explain the mechanisms of antiarrhythmic drug-action in ventricular arrhythmias.
Table 7–1.
Vaughan Williams classification of antiarrhythmic drug actions
I | Na+ channel block | Ia | Prolong APD | Quinidine Procainamide Ajmaline Disopyramide Cibenzoline Pirmenol |
Ib | Shorten APD | Lidocaine Mexiletine Tocainide Aprindine(no effect on APD) | ||
Ic | No effect on APD | Flecainide Pilsicainide Propafenone | ||
II | β-adrenergic receptor block | Propranolol Metoprolol Atenolol | ||
III | K+ channel block | APD prolongation | Amiodarone Bretylium Sotalol Dofetilide E-4031 | |
IV | Ca2+ channel block | Nifedipine Verapamil Diltiazem Bepridil | ||
Table 7–2.
Sicilian Gambit
Drug | Channels | Receptors | Pumps | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Na | Ca | K | If | α | β | M2 | P | Na/K ATPase | |||
Fast | Med | Slow | |||||||||
Lidocaine | ◯ | ||||||||||
Mexiletine | ◯ | ||||||||||
Tocainide | ◯ | ||||||||||
Moricizine |  | ||||||||||
Procainamide |  | ● | |||||||||
Disopyramide |  | ● | ◯ | ||||||||
Quinidine |  | ● | ◯ | ◯ | |||||||
Propafenone |  | ● | |||||||||
Flecainide |  | ◯ | |||||||||
Encainide |  | ||||||||||
Bepridil | ◯ |  | ● | ||||||||
Verapamil | ◯ |  | ● | ||||||||
Diltiazem | ● | ||||||||||
Bretylium |  | ◐ | ◐ | ||||||||
Sotalol |  |  | |||||||||
Amiodarone | ◯ | ◯ |  | ● | ● | ||||||
Alinidine | ● |  | |||||||||
Nadolol |  | ||||||||||
Propranolol | ◯ |  | |||||||||
Atropine |  | ||||||||||
Adenosine |  □ □ | ||||||||||
Digoxin |  □ □ |  ● ● | |||||||||
General Arrhythmia Suppression Mechanisms in Ventricular Arrhythmias
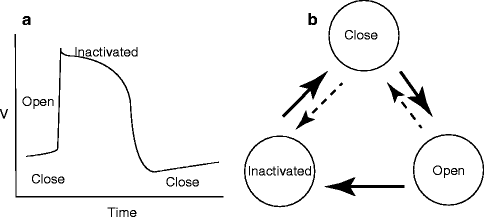
Antiarrhythmic drugs can block cardiac arrhythmias by suppressing underlying mechanisms, such as abnormal automaticity, delayed afterdepolarization (DAD), early afterdepolarization (EAD), and reentry (Fig. 7.2). Typically, the abnormal automaticity is caused by decrease of resting membrane conductance and/or enhancement of inward currents such as Ca2+ channel current. The DAD is due to the overload of intracellular calcium ions (Fig. 7.2a). EAD is caused by the excessive prolongation of APD (Fig. 7.2b). And Establishment of microreentry requires the unidirectional conduction block and slow conduction of AP (Fig. 7.2c). Abnormal automaticity can be altered by either changing the diastolic phase 4 slope, the threshold potential for rapid initial upstroke in phase 0, or APD. Generally speaking, Na+ channel blockers and Ca2+ channel blockers elevate the threshold potential for AP initiation, K+ channel blockers prolong the APD, adenosine hyperpolarizes the cell membrane potential and shorten the APD, and β-adrenergic receptor blockers decrease phase 4 slope. Similarly, Na+ channel blockers and Ca2+ channel blockers may suppress DAD by interfering with its upstroke. The EAD can be inhibited by shortening APD. Reentry may be terminated by prolonging the refractory period, i.e. by blocking delayed rectifier K+ channels or slowing recovery of Na+ channel from inactivation. The main purpose of this chapter is to describe the basic mechanisms of each class of drug affects cardiac ion channel activity.
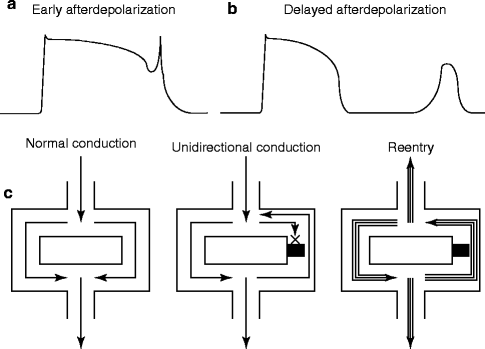
Figure 7–2.
(a) Early afterdepolarization (b) Delayed after depolarization (c) Normal conduction, unidirectional conduction and Reentry
Na+ Channel Blocker (Class I Drugs)
Class I drugs in the Vaughan-Williams classification are primary Na+ channel blockers and, historically, the blockers’ effect on the Na+ channel was first investigated to elucidate the mechanisms of antiarrhythmic drugs, before other antiarrhythmic drugs. Na+ channels in many excitable cells generate a rapid regenerative upstroke of action potential and the Na+ channel current is tetrodotoxin (TTX)-sensitive and/or μ-conotoxin sensitive. Two independent gating mechanisms of Na+ channel (activation and inactivation) give a rise to fast onset and decay of the Na+ channel current after depolarizing pulse (Fig. 7.3a). The Na+ channel in atria and ventricle accounts for the initial rapid depolarization (phase 0), which is responsible for the fast conduction of excitation in cardiac tissues. Its maximum upstroke slope (dV/dtmax) is approximately proportional to the Na+ channel current amplitude. And thus it has been used as the parameter to evaluate the effect of Class I drugs on the Na+ channel. Therefore, the primary effect of Class I drugs is to slow the upstroke of cardiac AP. Class I drugs are further divided into three subgroups (Ia, Ib, Ic), according to their electrophysiological effects on APD. For example, Class Ia drugs such as quinidine slow the maximum upstroke slope and in addition prolong APD (see Fig. 7.1b). Because of its APD prolongation effect, quinidine is also known to cause so-called quinidine-shock, i.e., quinidine-related torsades de pointes type of polymorphic ventricular tachycardia. On the other hand, Class Ib drugs, such as lidocaine, also make maximum upstroke slope gentle but shorten the APD (see Fig. 7.1b). Class Ic drugs slow the maximum upstroke slope but do not have a significant effect on APD. These differences among the Class I subtype drugs can be accounted for either by the kinetic properties of the drug action on Na+ channels and/or by action on other ion channels such as K+ channels.