Figure 8–1.
Factors involved in the initiation of atial fibrillation (AF) and pharmocological options to prevent the initiation of this arrhythmia
The maintenance of AF is facilitated by the development of both electrical and structural remodeling [10–12]. The electrical remodeling abbreviates ERP due to abbreviation of APD and the structural remodeling contributes to development of conduction disturbances [13]. Structural remodeling in atria also develops with age and is associated with a number of diseases, including heart failure, ischemic heart disease and hypertension.
Although AF occurrence is often associated with ERP shortening, the initiation of AF may not always be associated with abbreviation of ERP. In many pathologies associated with AF, such as atrial dilatation, heart failure and hypotension, atrial ERP may not be altered or may even be prolonged [14–18]. Interestingly, although age is a major risk factor for development of AF, ERP prolongation is paradoxically associated with advancing age in both dogs and humans [19, 20]. It seems that in many patients experiencing AF or having a history of AF, a short ERP is largely a consequence of AF itself. Indeed, during on-going AF or frequent episodes of AF, ERP appears to become short in any pathology.
An important concept in the science of AF generation is that of “wavelength”, defined as the product of ERP and conduction velocity. Simply defined, the wavelength is the length of the reentrant circuit occupied by the advancing wavefront and refractory to reexcitation. The value of the wavelength often predicts the probability of appearance/maintenance of AF (i.e., the shorter the wavelength the greater probability of AF induction and maintenance and vice versa) [21].
Anti-AF Pharmacology
The most common anti-AF approach involves prolongation of ERP, for both prevention initiation and termination of AF. Irrespective of the mechanisms underlying initiation or maintenance of AF, ERP prolongation is effective in slowing or terminating and/or preventing the re-induction of AF. Pharmacological prolongation of ERP can be achieved with prolongation of APD, induction of post-repolarization refractoriness (PRR), or by a combination of the two (Fig. 8.2). APD prolongation is largely due to inhibition of potassium current(s) and induction of PRR is exclusively due to block of the sodium channel responsible for peak INa. Multiple ion channel blockers can prolong ERP by both APD prolongation and induction of PRR.
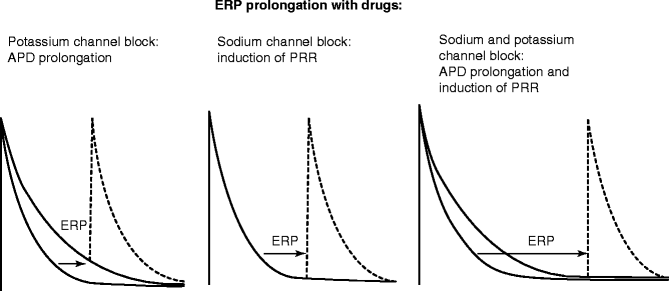
Figure 8–2.
Prolongation of atrial effective refractory period (ERP) with potassium and/or sodium channel blockers. This schematic depicts the effects of sodium, potassium and mixed ion-channel blockers on action potential duration and effective refractory period (ERP, depicted by arrows). APD action potential duration, PRR post-repolarization refractoriness
The role of conduction alterations in anti-AF action of INa blockers remains controversial. Conduction slowing/disturbance by itself is expected to promote reentrant arrhythmias by reducing wavelength. This, however, may be the case for the ventricles but not for atria because the use of INa blockers is associated with the induction of ventricular but not atrial fibrillation. For instance, at the same concentration range, pilsicainide promotes ventricular spiral wave-mediated arrhythmias [22], but suppresses spiral wave-induced AF [23]. This distinction appears to be due to a greater effect of sodium channel blockers to reduce excitability in atria vs. ventricles (discussed below), thus producing a much greater prolongation of ERP in atria. Improvement or normalization of conduction may exert an anti-AF action in some AF pathologies (like heart failure) [24]. Improvement of conduction in atria, however, remains difficult to achieve. In most AF cases, conduction slowing in atria is due to structural remodeling. While connexin (principal gap junction subunits) is significantly altered in atria of congestive heart failure dogs, these changes do not contribute to atrial conduction disturbances and AF in this setting [25]. Upstream therapy may be useful in preventing or ameliorating atrial structural remodeling and, thus, atrial conduction disturbances, but this remains to be determined [26].
AF prevention can be achieved by suppressing the trigger and by reducing the arrhythmogenic substrate (see Fig. 8.1). The initiation of AF is often thought to be caused by intracellular calcium mediated triggered activity/automaticity [27, 28]. Dynamic interaction between sympathetic and parasympathetic systems play a critical role in the initiation of paroxysmal AF [29]. In this context, intracellular-mediated late phase 3 EADs [30, 31] have been proposed to play a prominent role in the induction of AF [29, 31]. Pharmacological reduction or normalization of elevated intracellular calcium activity can suppress the development of calcium-mediated triggered activity and AF. This can be achieved with sodium and calcium channel blockers and, theoretically, with a modulator of autonomic nervous system activity [29, 31, 32]. The AF trigger can be suppressed not only by a direct effect (e.g., by a reduction/normalization of intracellular calcium or blocking a specific ion current causing DAD/EAD/automaticity), but also by a prolongation of ERP (simply moving the trigger beyond the AF vulnerable window; see Fig. 8.1).
Although a reduction of electrical heterogeneity is expected to decrease AF vulnerability, pharmacological reduction of electrical heterogeneity in atria and its utility in anti-AF action are poorly defined. It is noteworthy that the antiarrhythmic value of pharmacologically reducing dispersion of repolarization and refractoriness in the ventricle has been demonstrated in many arrhythmogenic pathologies, including the long QT, short QT, early repolarization and Brugada syndromes, catecholaminergic polymorphic ventricular tachycardia, heart failure, ischemia and infarction [33–37].
The electrophysiological efficacy of AADs to prolong ERP at regular and rapid activation rates can vary significantly (i.e., INa blockers are use-dependent and IKr blockers are reverse-use dependent). These properties should be reflected in anti-AF efficacy of IKr and INa blockers to prevent and terminate AF. Both potassium and sodium channel blockers have been shown to more readily prevent AF initiation than to terminate on-going AF [38–40]. These results are consistent with the reverse-use dependence of IKr blockers, but at first glance appear to contradict the use-dependent effects of INa inhibitors.
Sodium Channel Block for AF
Many antiarrhythmic agents that block INa as their primary action are classified as Class IA, IB or IC based on their unbinding kinetics from the sodium channel and their effect on APD in ventricular myocardium [41]. Class IB agents like lidocaine and mexiletine abbreviate APD and have rapidly unbinding kinetics from the sodium channel (τ < 1 s). Class IA agents, like procainamide, quinidine and disopyramide, prolong APD (largely due to block of IKr) and have intermediate unbinding kinetics (τ > 1 but <12 s). Class IC agents, like propafenone or flecainide, generally produce little to no effect on ventricular APD and manifest slow unbinding kinetics from the sodium channel (τ > 12 s). However, recently it became evident that propafenone may cause atrial-selective APD prolongation [42], like many other agents that inhibit IKr (amiodarone, ranolazine, AZD1305, E-4031, etc.) [43, 44]. While amiodarone is classified as Class III antiarrhythmic agents (prolonging APD), this agent potently blocks early INa as well (predominantly in atria vs. ventricles; with a rapid kinetics, corresponding to Class IB) [45, 46], contributing to anti-AF properties of this drug. Most of the “novel” anti-AF AADs (ranolazine, vernakalant, AZD1305, AVE0118 etc.) potently block peak INa (in an atrial-selective manner [44]), but have not been classified under the Vaughan Williams classification scheme.
A critical and unique feature of INa blockers related to their antiarrhythmic actions is their ability to produce rate-dependent PRR, i.e., to prolong ERP without APD prolongation or to a greater extent than APD prolongation. The effectiveness of most INa blockers to inhibit INa is typically enhanced following acceleration of activation rate, a phenomenon termed “use-dependence” [46, 47]. This property has long been recognized to be very useful for suppression of rapid arrhythmias (including AF), while causing a relatively small effect at normal activation rates. The rate dependence of INa blocker is related to a generally higher affinity of INa blockers for the open and/or inactivated state of the sodium channels (i.e., during the action potential) than to the rested channels (i.e., during the diastolic interval, when net unbinding occurs). Acceleration of heart rate increases the proportion of time during which the sodium channels are in open and/or inactivated states vs. rested state. The efficacy of sodium channel blockade is normally augmented by depolarization of resting membrane potential (RMP), due to increases in the fraction of inactivated vs. rested sodium channels as well as slowed unbinding kinetics at these more positive potentials. APD shortening tends to reduce the efficacy of sodium channel blockade due to a relative decrease of the time during which the sodium channels remain in the inactivated state vs. rested state [45–47].
In the clinic, Class IC and Class IA (which also reduce IKr), but not Class 1B (relatively selective INa blockers) agents can effectively suppress paroxysmal AF. Class IA agents, however, are rarely used because of the risk of induction of TdP due to delayed ventricular repolarization [48]. Due to absence of “pure” INa blockers with slow/medium kinetics, it remains unknown whether a “pure” INa block is capable of effectively suppressing clinical AF. Note that pilsicainide, a purported highly selective early INa blocker with slow kinetics (a Class IC agent, used in Japan [49]) inhibits IKr and may cause long QT [50]. Very high (toxic) concentrations of “pure” INa blockers (lidocaine and TTX) can be very effective in preventing/terminating AF in experimental and theoretical settings because of their effects to reduce excitability at these high levels of INa inhibition [51, 52].
The antiarrhythmic mechanisms underlying anti-AF efficacy of INa blockers is multi-factorial, involving rate-dependent prolongation of ERP (largely due to PRR), depression of excitability, impaired impulse propagation, as well as non-INa -mediated influences, i.e., the prolongation of APD, due to inhibition of IKr. It is important to recognize that APD prolongation can significantly promote block of INa, secondary to reducing diastolic interval, during which much of the recovery from block occurs, and a more positive take-off potential, both resulting in reduced availability of sodium channels. The development of PRR secondary to reduced excitability appears to be the most important anti-AF action of INa blockers (Figs. 8.2 and 8.3) [39, 42, 43, 53, 54]. Inhibition of both peak and late INa also contributes to suppression of DADs, EADs, and automaticity, which contribute to the anti-AF action of the sodium channel blockers [55, 56].
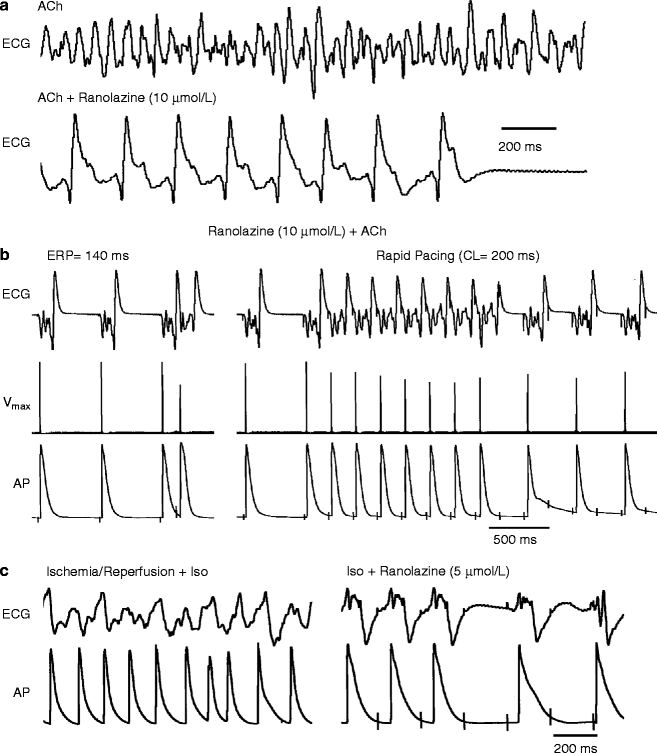
Figure 8–3.
Ranolazine suppresses AF and/or prevents its induction in two experimental models involving isolated canine arterially-perfused right atria. (a) Persistent ACh (0.5 μM)-mediated AF is suppressed by ranolazine (10 μM). AF initially converts to flutter and then to sinus rhythm. (b): ERP measured at a CL of 500 ms is 140 ms. Attempts to re-induce AF fail because ranolazine-induced depression of excitability leads to 1:1 activation failure soon after CL is reduced from 500 to 200 ms (right panel). (c) Rapid-pacing induces non-sustained AF (48 s duration) following ischemia/reperfusion plus isoproterenol (Iso, 0.2 μmol/L) (left panel). Ranolazine (5 μM) prevents pacing-induced AF due to 1:1 activation failure (right panel). In both models, ranolazine causes prominent use-dependent induction of post-repolarization refractoriness (Reproduced with permission from Burashnikov et al. [39] with kind permission from Wolters Kluwer Health)
It has long been appreciated that INa blockers (Class IC) can effectively prevent and terminate paroxysmal AF, but not persistent AF [57]. This may be due to electrical remodeling causing atrial APD abbreviation as well as to significant structural remodeling, which is observed in patients with long-enduring AF [58]. Structural remodeling can contribute to conduction slowing/disturbances. It is unclear if the ability of INa blockers to inhibit peak INa is reduced in persistent AF. The efficacy of Class IC agents to depress INa -mediated parameters remains well preserved in remodeled atria of goats [59, 60]. Class IC drug-induced prolongation of AF cycle length needed for termination of persistent AF becomes progressively longer with continuation of AF [59].
INa blockers that dissociate rapidly form the sodium channels (e.g., ranolazine, chronic amiodarone and AZD1305) have been shown to produce atrial-selective depression of sodium channel-dependent parameters and to effectively suppress AF in canine coronary-perfused atrial preparations at concentration that cause little or no effect in the ventricles [39, 43, 54, 61–63]. The atrial selectivity and anti-AF efficacy of ranolazine, AZD7009, and AZD1305 have been demonstrated in porcine and canine hearts in vivo and in vitro [54, 61, 62]. Vernakalant also appears to be an atrial-selective INa blocker since it slows conduction velocity in atria, but not in the ventricles and prolongs atrial ERP largely due to induction of PRR [64, 65].
Dronedarone, an amiodarone derivative, causes atrial but not ventricular PRR in canine and porcine hearts [40, 66]. The dronedarone-induced PRR is much less than that induced by amiodrone. Dronedarone’s inhibition of peak INa is very voltage-dependent in guinea pig ventricular myocytes [66], which may contribute to dronedarone’s atrial-selective induction of PRR, owing to the fact that the resting membrane potential of atrial cells is more depolarized than that of ventricular cells.
A number of factors underlie the atrial-selective effects of INa blockers, including a more negative steady-state inactivation relationship, a more positive resting membrane potential (RMP), and a more gradual phase 3 of the action potential in atrial vs. ventricular cells [39, 67–69]. The more negative half-inactivation voltage and more positive RMP importantly reduce the fraction of resting channels in atria vs. ventricles at RMP. Because recovery from sodium channel block occurs predominantly during the resting state of the channel, accumulation of sodium channel block is expected to be greater in atria vs. ventricles.
There is significant variability in the degree to which sodium channel blockers are atrial-selective [67, 68, 70]. Available data suggest that binding affinity of the INa blocker for a given state of the channel (i.e., open, inactivated, or resting) does not determine the drug’s atrial-selectivity (for review see [67, 71]). The rate of dissociation of the drug from the sodium channel, however, appears to be key. INa blockers possessing rapid vs. slow unbinding kinetics tend to be highly atrial-selective (e.g., ranolazine, vernakalant, chronic amiodarone, but not propafenone) [42, 67, 69]. Agents like flecainide and propafenone, which dissociate relatively slowly from the sodium channel, allow for accumulation of block in both atria and ventricles at rapid rates, leading to absence of atrial-selectivity and in some cases ventricular-predominant effects [42, 72].
Atrial selective INa blockers have been shown to be effective in the management of AF in the clinic. A number of clinical studies have demonstrated the ability of ranolazine to prevent the induction of AF and terminate paroxysms of AF using a “pill-in-the-pocket” approach [73–76]. AZD7009 and AZD1305 have been reported to effectively suppress clinical AF [77–79]. Amiodarone is the best available agent for the long-term maintenance of sinus rhythm in AF patients. All of these agents are atrial-selective INa blockers (ranolazine, amiodarone, AZD7009, and AZD1305), that also inhibit other ion channels (particularly IKr), which is likely to importantly contribute to their atrial selectivity and anti-AF efficacy (discussed below). The degree to which the clinical efficacy of these AADs depends on block of INa remains to be determined. Of note, lidocaine, a “mild” atrial selective INa blocker [39] is not particularly effective against clinical AF, presumably because it is a relatively “pure” INa blocker.
Potassium Channel Block for AF
The common functional manifestation of block of potassium channels is the prolongation of APD90 and, thus, lengthening of ERP (see Fig. 8.2). The most prominent clinically- proven anti-AF potassium channel blockers are those that inhibit the rapidly activating delayed rectified potassium current (IKr; such as dofetilide, sotalol, ibutilide). Anti-arrhythmic agents causing prolongation of APD are commonly classified as the Class III agents. Anti-AF efficacy of the slowly activating delayed rectified potassium current (IKs) has also been reported [80, 81], but remains poorly defined. It appears that the anti-AF effectiveness of IKs block is poor, but that it can enhance the anti-AF efficacy of IKr inhibition [81]. It is noteworthy that many multiple ion channel blockers inhibit IKs (e.g., amiodarone and quinidine).
There has been great interest in recent years in the anti-AF effectiveness of inhibition of atrial-specific potassium channels, including the channels that carry the ultra-rapid delayed rectifier potassium current (IKur), the acetylcholine-regulated inward rectifying potassium current (IK-ACh), and the constitutively active IK-ACh (i.e., which does not require acetylcholine or muscarinic receptors for activation) [44, 82].
Block of IK-ACh or constitutively active IK-ACh may suppress vagally-mediated AF or AF in which vagal activity contribute to the initiation of paroxysmal AF. Clinical data indicate that vagal components contribute to the initiation of paroxysmal AF [32, 83]. Block of IK-ACh currents with tertiapin-Q prolongs atrial APD and suppresses AF in experimental models [84, 85]. Interestingly, CA-IKACh is only marginally present in healthy non-fibrillating human or canine atria and is significantly increased in atria of chronic AF patients and canine tachycardia-remodeled atria [84, 86–88], suggesting that this current is a pathology-specific as well as atrial-specific target [89]. At present, a CA-IKACh–selective blocker is not available.
IKur is the most investigated atrial-specific ion channel current and until recently was widely considered to be the most promising among atrial-specific targets for the treatment of AF [82, 90]. Enthusiasm for specific IKur blockers for the management of AF has diminished in recent years [67, 91–94]. Inhibition of IKur alone abbreviates APD and ERP in “healthy” atrial cells and produces only a minor ERP prolongation in remodeled atrial cells. Available data indicate that inhibition of IKur alone is ineffective against AF [91, 93, 94]. In fact, all prominent Ikur blockers (e.g., vernakalant, AZD1305 and AVE0118) inhibits peak INa and their atrial selective ERP prolongation is largely or exclusively due inhibition of peak INa, not IKur [44]. The apparent failure of pure IKur inhibition for suppression of AF may be explained by several factors. First, the contribution of IKur during AF is likely to be relatively small because IKur density is reduced with acceleration of activation rate [95]. Second, the contribution of IKur to atrial repolarization may be reduced in AF patients, since IKur density is reported to be decreased in cells isolated from atria of patients with chronic AF [96, 97].
Among the potassium currents, IKr block is the only one that has been proven to be widely effective against AF in the clinic. It is of some interest that all clinically-effective INa blockers also inhibit potassium currents (primarily IKr).
IKr blockers inhibit the channel in its open state, i.e., during phases 2 and 3 of the action potential. As a consequence, the inhibiting efficacy of IKr blockers is importantly determined by the pre-drug APD, i.e., the shorter pre-drug APD the smaller APD prolongation induced by IKr block and vice versa. The efficacy of IKr blockers to prolong APD is also rate-dependent, i.e., the faster the rate the weaker the efficacy. Thus, IKr block-induced APD prolongation is the smallest at rapid activation rates, where ERP prolongation is most desirable for AF suppression. This apparent disadvantage of IKr blockers for suppression of cardiac arrhythmias has been long recognized [98, 99].
Prolongation of ERP secondary to prolongation of AP is the primary anti-AF mechanism of IKr and other K channel blockers (see Fig. 8.2). Indeed, ERP prolongation has been associated with prevention and termination of AF in a vast majority cases with IKr blockers [100]. The ability of IKr blockers to prolong APD/ERP is decreased in remodeled atria in humans as well as in animal models with persistent AF (where APD is commonly short) [60, 101]. This may account for or contribute to a reduction of success rate of IKr inhibition to terminate persistent AF [57]. Still, IKr block using dofetilide or ibutilide remains the best available pharmacological approach for termination of persistent AF [57].
Multiple Channel Blockers for AF
With the exception of selective IKr blockers, such as dofetilide and sotalol, all clinically effective anti-AF agents inhibit multiple ion channels, principally peak INa and IKr (e.g., amiodarone, flecainide and propafenone). Recent studies have highlighted the importance of the ability of INa blockers to also block IKr and prolong the atrial AP. IKr block produces atrial-predominant prolongation of ERP/APD [43, 102], which significantly promotes block of peak INa particularly at rapid activation rate (Fig. 8.4). This translates into a greater development of atrial-selective PRR. Atrial-selective ERP prolongation is a desirable property of anti-AF drugs, which reduces the probability of proarrhythmia in the ventricles [69, 103].
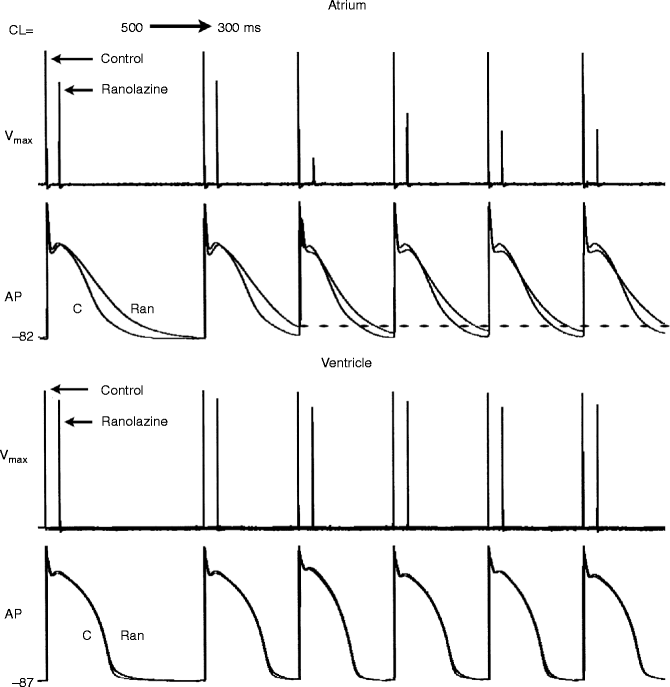
Figure 8–4.
Ranolazine produces a much greater rate-dependent inhibition of the maximum action potential upstroke velocity (V max ) in atria than in ventricles. Shown are Vmax and action potential (AP) recordings obtained from coronary-perfused canine right atrial and left ventricular preparations before (C) and after ranolazine (10 μM). Ranolazine prolongs AP duration in atria, but not the ventricles (due to IKr inhibition [43]). Acceleration of rate leads in the presence of ranolazine leads to a more positive take-off potential and elimination of the diastolic interval. Because much of the recovery from sodium channel block occurs during the diastolic interval, both effects lead to a reduced availability of sodium channels. Loss of the diastolic interval in atria but not in the ventricle accounts for the atrial selective depression of sodium channel current as signified by Vmax (Reproduced from Antzelevitch and Burashnikov [71], with kind permission from Elsevier)
Prolongation of APD leads to a more depolarized take-off potential as well as loss of the diastolic interval at rapid rates of activation. Both effects contribute to reduced availability of sodium channels for activation. Prolongation of APD is especially effective in potentiating the effect of INa blockers that dissociate rapidly from the sodium channel, such as amiodarone and ranolazine (Fig. 8.4). This synergism of combined INa and IKr block forms the basis for the high anti-AF efficacy of these multi-ion channel inhibitors. Rapid dissociation of INa blockers from the sodium channel also contributes to their atrial selectivity, because ventricular tissues maintain their diastolic intervals at rapid rates, allowing dissociation of the drug before the next beat. The loss of the diastolic interval in atria slows drug dissociation, thereby allowing for the accumulation of block beat to beat [67].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


