Fig. 19.1
Main clinical features of Marfan syndrome. (a) Aortic root dilatation; (b) ectopia lentis; (c) skeletal abnormalities
In the absence of a family history of MFS, the diagnosis is confirmed when:
with Ao (Z ≥ 2) as aortic root dilatation, defined as a diameter of the aorta at the level of the sinuses of Valsalva exceeding two standard deviations (Z ≥ 2) above the mean; EL as ectopia lentis; Syst as systemic score, consisting of a list of features in different organ systems (see reference); and FBN1 as documented mutation in the FBN1 gene.
1.
Ao (Z ≥ 2) + EL = MFS
2.
Ao (Z ≥ 2) + FBN1 = MFS
3.
Ao (Z ≥ 2) + Syst (≥7 pts) = MFS
4.
EL + FBN1 with known Ao = MFS
In the presence of a family history (FH), criteria are slightly less strict:
5.
EL + FH of MFS (as defined above) = MFS
6.
Syst (≥7 pts) + FH of MFS (as defined above) = MFS
7.
Ao (Z ≥ 2 in adults, Z ≥ 3 in children) + FH of MFS (as defined above) = MFS
MFS shows virtually complete penetrance but the clinical spectrum is highly variable, even within families. Modifiers of the clinical severity are largely unknown. Up to 30 % of cases are due to new dominant mutations.
To date, there are no reproducible genotype-phenotype correlations in MFS with the exception of the neonatal form being caused by mutations in the middle region of the gene (exons 24–32) [3]. Neonatal Marfan syndrome is the most severe form of the disorder, with death occurring early in life, often during the first year.
19.1.2 Clinical Cardiovascular Manifestations
19.1.2.1 Aortic Disease
Most of the morbidity and mortality associated with MFS is related to the cardiovascular manifestations. The most common lesion is aortic root dilatation leading to aneurysm formation, which can be complicated by aortic dissection or rupture. It is estimated that aortic root dilatation is present in >80 % of adult MFS patients [1]. In surgical series, up to one-third of MFS patients present with aortic dissection, mainly Stanford type A dissection, indicating the persisting deficiency of timely diagnosis and adequate risk estimation [4].
The predilection for the ascending aorta to dilate is a result of both structural and local haemodynamic factors. Both the aortic root and the proximal part of the pulmonary artery are derived from the neural crest, whereas the more distal arterial structures stem from the mesodermis. It has been demonstrated that the elastic fibre content is higher in the ascending aorta than in any other region of the arterial tree [5]. Diseases such as MFS affecting elastic fibre integrity will therefore manifest more easily in this region. Furthermore, it is primarily the ascending portion of the aorta which is subjected to the repetitive stress of left ventricular ejection, eventually resulting in progressive dilatation [6, 7]. Since pressures in the aorta are significantly higher than in the pulmonary artery, dilatation will be more pronounced at the aortic root. Pulmonary artery dilatation occurs commonly in MFS but does not carry the same risk of dissection [8–10].
Up to now, complications in the descending aorta are limited to a minority of MFS patients, but the incidence of descending thoracic aortic dissection may increase since medical and surgical treatments of ascending aortic complications have significantly improved, hence leaving the descending aorta at risk [11, 12]. Marfan syndrome patients having thoraco-abdominal aortic aneurysm/dissection as a presenting manifestation are reported in a few case reports [13, 14], and it is notable that dissection in the descending part of the aorta is independent of the diameter of the ascending aorta [15]. Other data on the descending aorta in MFS patients are found in surgical reports describing the occurrence of primary or secondary complications in the descending aorta necessitating surgical intervention. Eight to fifteen percent of Marfan patients require initial surgery in the descending aorta [4, 11]. Patients with initial type B aortic dissection are at a significant higher risk for re-intervention (86 % for previous type B dissection versus 42 % for previous type A dissection). The majority of re-interventions are required in patients with previous dissection (48 % versus 11 % re-intervention in the patients presenting with aortic aneurysm) [4].
Altered anatomical features in the aorta of MFS patients, such as dilatation and/or dissection, are accompanied by functional impairment of the vessel, as reflected in increased aortic stiffness in MFS patients. Abnormal elastic properties – pulse wave velocity and local aortic distensibility – are not confined to the ascending aorta but are also detected in the normal-sized, more distal parts of the vessel [16, 17], and this as well in patients having previously undergone aortic root surgery as in unoperated MFS patients [18]. Interestingly, aortic stiffness seems to be an early marker of aortic disease as patients without thoracic aortic dilatation already demonstrate significant decreased aortic distensibility at proximal as well as distal levels of the aorta [17]. Local distensibility of the descending thoracic aorta appeared to be an independent predictor of progressive descending aortic dilatation [19].
19.1.2.2 Mitral Valve Prolapse
Mitral valve prolapse is another common cardiovascular complication of the Marfan syndrome occurring in up to 80 % of patients [10, 20, 21]. It is unclear whether the prevalence of MVP is increasing with age. In a large paediatric cohort, MVP occurred more commonly in females, but this female preponderance was not confirmed in adult series. A recent study showed that MFS patients have an increased risk of 28 % for MV-related clinical events at a younger age (endocarditis, surgery and heart failure) versus 13 % in idiopathic MVP [22]. Five to twelve percent of MFS patients will require mitral valve surgery as a primary cardiovascular intervention [23].
19.1.2.3 Cardiomyopathy and Arrhythmias
Although not commonly acknowledged in MFS, dilated cardiomyopathy, beyond that explained by aortic or mitral valve regurgitation, seems to occur with a higher prevalence, suggesting a role for the extracellular matrix protein fibrillin-1 in the myocardium. Significant LV dilatation and dysfunction leading to heart failure and necessitating heart transplantation has been described in a few cases and seems to be a very rare complication. Subclinical myocardial dysfunction on the other hand has been reported in larger subsets of MFS patients of various ages by several independent research groups [24–29], and mildly increased LV dimensions have been demonstrated in a subset of patients with MFS [30]. A study assessing genotype-phenotype correlations indicated that LV dilatation was more frequently observed in patients with a non-missense FBN1 mutation [31].
19.1.3 Aetiology and Pathophysiology
The pleiotropic clinical manifestations observed in MFS strongly suggested that the causal factor for the disease had to be found in a widespread tissue component such as the connective tissue. Applying monoclonal antibody studies in mice, Hollister, Sakai and their co-workers discovered a putative candidate, localized to numerous tissues including the aortic media and ciliary zonule, with the fibrils visualized termed fibrillin [35]. Fibrillin fibrils are a part of the 10 nm elastin-associated microfibrils. Immunohistochemical studies were the first to suggest that fibrillin is implicated in MFS [36]. The ultimate proof had to await the discovery of the gene encoding fibrillin (the fibrillin-1 gene, FBN1) and subsequent genetic linkage and mutation studies [37, 38]. To date more than 1,000 different mutations have been identified throughout the gene. Most mutations are unique to that individual or family.
The histological hallmark in the aorta of Marfan syndrome is the so-called cystic medial degeneration, with degeneration of the elastic fibres, irregular hypertrophy and apoptosis of vascular smooth muscle cells and increased basophilic ground substance within cell-depleted areas.
Conventional knowledge held that aneurysm formation in MFS results from an inherent structural weakness of connective tissues. Studies in different mouse models for MFS have extended this knowledge by demonstrating that fibrillin-1 also plays an important functional role in regulating transforming growth factor-beta (TGFβ) bioavailability. TGFβ is a multifunctional peptide that controls proliferation, differentiation and other functions in many cell types. Sakai and co-workers demonstrated that fibrillin-1 is homologous to the family of latent TGFβ-binding proteins (LTBPs), which serve to hold TGFβ in an inactive complex in various tissues, including the extracellular matrix [39], and it was known that fibrillin-1 binds TGFβ and LTBPs [40–42]. Increased TGFβ signalling has been demonstrated in several tissues in MFS patients.
More recent studies by Charbonneau and colleagues demonstrated that an Fbn1 mouse in which the LTBP binding site was deleted (Fbn1 H1Δ ) did not present features of MFS [43]. Hence, instead of reduced TGFβ sequestration, mutant microfibrils probably influence TGFβ activation in a different way. More recent reports now suggest that this increased signalling is the result of a final common pathway in the disease process and that the role of the TGFβ signalling pathway may vary during the dynamic transition from aortic aneurysm predisposition to end-stage disease, such as dissection [44]. A recent and very interesting hypothesis states that in case of loss of extracellular matrix integrity by mutated microfibrils, mesenchymal cells, such as fibroblasts or smooth muscle cells, detect the defective matrix and respond by repairing the failed matrix through the generation of active TGFβ and production of the required activators of latent TGFβ as part of the repair process [45]. Other mechanisms such as mitochondrial dysfunction and compromised VSMC differentiation may also play an important role. A schematic overview of the pathogenesis is provided in Fig. 19.2.
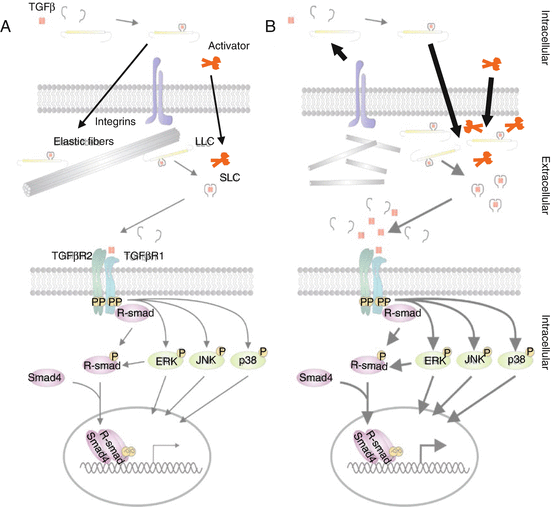
Fig. 19.2
Pathogenic mechanism of Marfan syndrome. (a) TGFβ signalling in healthy individuals. (b) Fragmented microfibres lead to a damaged ECM and initiate repair responses producing more (enhanced arrows) ECM constituents, SLCs and LLCs together with their activators; subsequently, enhanced (bold arrows) signalling through canonical (purple) and non-canonical (green) TGFβ signalling results in changes in target gene expression in MFS patients
The processes occurring in the aortic ECM due to mutated FBN1 are further influenced by altered haemodynamic stress in the aorta. As such, mechano-transduction of haemodynamic stress further exacerbates the tissue-related activation of vascular smooth muscle cells, matrix metalloproteinases and TGFβ.
19.1.4 Management and Treatment of Marfan Syndrome
The pleiotropic nature of Marfan disease implies a multidisciplinary approach and appropriate treatment, involving among others ophthalmologists, physiotherapists, orthopaedic surgeons, cardiologists and cardiac surgeons. In this chapter, we will focus on the treatment of aortic disease, which mainly determines life expectancy in MFS and has been a main clinical and research focus for many decades. Improved diagnosis and better medical and surgical treatment have already resulted in a spectacular increase in life expectancy of more than 30 years [46] – no other cardiovascular disease has seen a similar improvement. The cornerstone of adequate management in MFS is correct and timely diagnosis, necessitating adequate education of all involved healthcare workers and correct echocardiographic assessment of the aortic dimensions by (paediatric) cardiologists according to established guidelines [47]. CT or MRI can be used in case of insufficient visualization of the ascending aorta by echocardiography. Once the diagnosis is confirmed, a first reassessment of the aortic dimension is recommended after 6 months to assess evolutionary changes. Further follow-up is guided by the diameter, evolution, underlying diagnosis and family history. Stable diameters <45 mm in MFS patients without a family history of dissection require yearly follow-up. Biannual controls are recommended in all other cases. Despite rigorous follow-up and adherence to the guidelines, some MFS patients will still succumb to (fatal) aortic dissection, indicating the need for improved risk stratification. Ongoing studies are identifying potential circulating biomarkers, including fibrillin-1 fragments, which may be used in conjunction with imaging data to better identify patients at risk for aortic dissection [48].
The ultimate goal for the treatment of aortic complications in MFS is to obtain a reduction in the occurrence rate of (fatal) aortic dissection, and it is beyond any doubt that – up to now – prophylactic aortic root surgery is the most successful preventive intervention. Refinements of surgical techniques, mainly with the introduction of valve-sparing procedures, have led to excellent short- and middle-term results and have obviated the need for chronic use of anticoagulants [49]. Surgery is routinely recommended in MFS patients when the diameter of the aortic root attains 50 mm. A reduced threshold of 45 mm is warranted in cases of rapid growth (>0.2-0.5 cm/year), cases with a positive family history of aortic dissection and/or significant aortic valve regurgitation and in women with desire of pregnancy [50, 51].
Since aortic surgery is inherently associated with a (small) risk for complications and a prolonged rehabilitation period, attempts have been made to at least postpone the need for surgery. Ultimately, arresting aortic growth and, conceptually, interference with the underlying genetic defect are deemed to be the ideal solution, which is – alas – not (yet) feasible at the moment.
Slowing down aortic root growth may be achieved through reducing haemodynamic stress on the proximal aorta. The first report on the use of β-blockers in MFS dates from 1971 indicating that reduction in the rate of increase in aortic pressure over time (dP/dt) was more effective than could be explained by reduction of blood pressure alone [52]. Subsequent small studies with β-blockers in turkeys that are prone to aortic dissection and in uncontrolled human studies of MFS had varying results [53, 54].
Since then, no less than 1,583 Marfan patients have been included in at least 19 different trials since the late 1970s [6, 53, 55–71]. Only one trial, showing a reduction in aortic root growth with propranolol, had a placebo-controlled randomized design [57]. Many other trials have confirmed the effect of β-blockers on aortic growth, and in some trials, beneficial effects on the elastic properties of the aorta have been reported [58]. This notwithstanding, the beneficial effect of β-blockers has not been consistent in all studies. Differences in the populations studied, in the drug types and dosage and in the study design, render the interpretation and comparison of these trials particularly challenging.
Another aspect that remains elusive despite the numerous trials is the timing of initiation of treatment – some advocate treatment as soon as the diagnosis is made while others suggest to wait until some dilatation has occurred.
Alternatives for β-blockers have been suggested and studied in small series.
Calcium channel blockers have been investigated in a very small study [59], showing similar results as β-blockers. ACE inhibitors (ACEIs) were postulated to have therapeutic value in MFS because of their potential ability to block apoptotic pathways through inhibition of the AT2 receptor. An open-label, nonrandomized trial of ACEIs versus β-blockers in MFS showed apparent therapeutic value for ACEIs in terms of reduced aortic stiffness and smaller increases in aortic root diameter [61]. Another randomized trial of perindopril versus placebo in MFS revealed improved biomechanical properties of the aorta, slower aortic root growth and lower levels of circulating TGFβ [64].
A seemingly major breakthrough in the search for improved medical treatment in MFS patients was achieved with the documentation of the involvement of the TGFβ pathway in the process of aneurysm formation. This led to the insight that interference with TGFβ signalling may have a beneficial impact on aortic growth. Losartan, an ARB with known TGFβ-inhibiting potential, was tested in a mouse model for MFS showing normalization of aortic root growth and restoration of elastic fibre fragmentation [72]. Human trials published so far may suggest a beneficial effect of the combination of beta-blocker with losartan treatment [67, 70]. Very recently, the results of a large randomized trial in a young (1mo-25y) MFS cohort, conducted by the Pediatric Heart Network comparing Atenolol and Losartan were published showing no difference in aortic root growth rate between both groups [73].
19.2 Associated Thoracic Aortic Aneurysm Syndromes
Some patients and families do present with thoracic aortic disease but do not fulfil the diagnostic criteria for MFS and/or present with other clinical features that do not fit within the MFS spectrum. Moreover, since the more widespread availability of molecular genetic testing in MFS, it became clear that subgroups of patients and families exist that do not carry an FBN1 mutation, suggesting the involvement of other genes.
Over the past decade, it has indeed been evidenced that heritable thoracic aortic disease (H-TAD) is genetically heterogeneous with a dozen genes being identified so far [74]. Most of these genetic conditions transmit as an autosomal dominant trait. Genes may be subdivided in three major categories: (1) genes involved in the extracellular matrix (FBN1, COL3A1, FBNL4), (2) genes involved in the TGFβ pathway (TGFBR1 and TGFBR2, SMAD3, TGFβ2, SKI) and (3) genes involved in the vascular smooth muscle cell apparatus (ACTA2, MYLK, MYH11, PRKG1).
For most of these genes, both syndromic and non-syndromic disease entities have been described. Though some clinical features may be highly discriminative (e.g. lens luxation for MFS), there is substantial clinical overlap between these genetic entities. Molecular genetic testing is a helpful additional tool in many cases, not only for diagnostic purposes but also because medical management may vary according to the underlying condition.
The main clinical entities with their respective genes and clinical features are listed in Table 19.1. A brief description of the most notable clinical entities in the context of arterial disease is provided below.
Table 19.1
Schematic overview of syndromic and non-syndromic TAAD (thoracic aortic aneurysm and dissections)
Disorder | Gene(s) | Main cardiovascular features | Additional clinical features | |
|---|---|---|---|---|
Syndromic H–TAD | ||||
FBN1 | Aortic root aneurysm, aortic dissection, mitral valve prolapse, main pulmonary artery dilatation, ventricular dysfunction | Lens luxation, skeletal features (arachnodactylia, pectus deformity, scoliosis, flat feet, increased arm span, dolichocephalia) | ||
Vascular Ehlers-Danlos [75] | COL3A1 | Aortic and major branching vessel dissection/rupture, often without preceding dilatation | Thin, translucent skin, dystrophic scars, facial characteristics (Madonna face, thin lips, deep-set eyes) | |
TGFβ-related vasculopathies | TGFBR1/TGFBR2 | Aortic root aneurysm, aortic dissection, arterial aneurysms and dissections, arterial tortuosity, mitral valve prolapse, congenital cardiac malformations | Bifid uvula/cleft palate, hypertelorism, pectus abnormalities, scoliosis, club feet | |
SMAD3 | Aortic root aneurysm, aortic dissection, arterial aneurysms and dissections, arterial tortuosity, mitral valve prolapse, congenital cardiac malformations | Osteoarthritis, soft skin, flat feet, scoliosis, recurrent hernias, hypertelorism, pectus abnormalities | ||
TGFβ2 | Aortic root aneurysm, aortic dissection, arterial aneurysms and dissections, arterial tortuosity, mitral valve prolapse, congenital cardiac malformations | Club feet, soft translucent skin | ||
SKI | Mild aortic root dilatation, mitral valve prolapse | Craniosynostosis, distinctive craniofacial features, skeletal changes, neurologic abnormalities, mild-to-moderate intellectual disability | ||
Non–syndromic H–TAD | ||||
TGFBR1/TGFBR2 (3–5 %) | Thoracic aortic aneurysm/dissection | Lack of syndromic features | ||
ACTA2 (10–14 %) | Thoracic aortic aneurysm/dissection, BAV cerebrovascular disease, coronary artery disease | Lack of marfanoid skeletal features, livedo reticularis, iris flocculi | ||
MYLK | Thoracic aortic aneurysm/dissection | Gastrointestinal abnormalities | ||
SMAD3 (2 %) | Intracranial and other arterial aneurysms | |||
TGF β 2 | Mitral valve prolapse | |||
PRKG1 | Thoracic aortic aneurysm/dissection, arterial aneurysms and dissections, arterial tortuosity | |||
MYH11 | Patent ductus arteriosus | |||
The identification of new genes in H-TAD has offered important new opportunities for the diagnosis and gene-tailored management of patients and families. In addition, these new genes have offered us unique new insights into the pathogenesis of aneurysmal disease. Identification of mutations in the various components of the ECM, the TGFβ pathway and the contractile unit of vascular smooth muscle cells provide the ultimate proof for their involvement into the process of aneurysm formation as described above.
19.2.1 Loeys-Dietz Syndrome
Loeys-Dietz syndrome (LDS) is caused by mutations in either of the transforming growth factor-beta receptor genes (TGFBR1 and TGFBR2). In its most typical form, LDS is characterized by aortic aneurysms and dissections, widespread arterial tortuosity/aneurysms, bifid uvula and hypertelorism (wide-spaced eyes) [76].
The cardiovascular lesions in LDS are more widespread, extending beyond the aorta, and may have a more aggressive course than in MFS with a higher annual increase in aortic root diameter in at least a subset of patients [77]. The expression of the disease is highly variable though, both between and within families. Milder presentations and even non-penetrance has been observed [90]. Larger-scale prospective registries are required to gain more insight into the course and presentation of the disease. Since aortic dissection has been reported in patients with aortic diameters well below conventional surgical thresholds, aortic root surgery in LDS is recommended at a diameter of 43–45 mm [50].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


