Management of Supraventricular Arrhythmias in Patients with Heart Failure
Gregory K. Feld
Doug Gibson
David Krummen
Heart failure is associated with significant morbidity and mortality. Cardiac arrythmias are particularly common in patients with this condition and they appear to increase risk for an event. The greatest risk of mortality is associated with ventricular arrhythmias, which can lead to sudden death. However, supraventricular arrhythmias may also increase morbidity and mortality in heart failure patients (1,2). Of particular concern is the development of atrial fibrillation, which may lead to systemic emboli causing stroke and increased risk of mortality (3,4,5). Due to the complex mechanisms of supraventricular arrhythmias (Table 40-1) and the potential adverse effects of standard pharmacological therapy in patients with heart failure (Table 40-2), the approach to their diagnosis and treatment (Table 40-3) represents a formidable challenge to the clinician. The diagnosis and management of supraventricular arrhythmias in the patient with heart failure will be addressed in this chapter.
Pathophysiology of Heart Failure as it Relates to Development of Supraventricular Arrhythmias
Supraventricular arrhythmias are more likely to occur in heart failure patients as a result of the associated hemodynamic and electrophysiological abnormalities. Heart failure is typically characterized by increased ventricular diastolic and atrial pressure and, not infrequently, mitral and tricuspid valve regurgitation—abnormalities that may ultimately lead to atrial dilatation and hypertrophy and fibrosis. These hemodynamic and pathophysiological derangements may then lead to abnormalities in atrial electrophysiology, a process called electrical remodeling, with resultant shortening and dispersion of atrial refractoriness and depression of conduction velocity, all of which may predispose to the development of supraventricular arrhythmias (6,7).
Patients with heart failure may also have an abnormally low cardiac output and a compensatory increase in sympathetic nervous system tone, which further increases the risk for development of supraventricular arrhythmias and may make them even more difficult to treat pharmacologically. In addition, since many of the antiarrhythmic drugs used to treat atrial arrhythmias have negative inotropic and chronotropic effects (i.e., they depress contractility and heart rate, respectively), they may worsen heart failure, which, in turn, may reduce their efficacy. Thus, heart failure with its associated hemodynamic and pathophysiological abnormalities creates a favorable electrophysiological milieu the development of supraventricular arrhythmias and complicates their treatment.
Diagnosis and Treatment of Supraventricular Arrhythmias
Paroxysmal Supraventricular Tachycardia
Supraventricular arrhythmias of any type (Table 40-1), including paroxysmal supraventricular tachycardia (PSVT), may occur in patients with heart failure. The most common forms of PSVT in the general population (8,9,10) include atrioventricular (AV) nodal re-entrant tachycardia (AVNRT) due to dual AV nodal physiology (Fig. 40-1A,B) and atrioventricular re-entrant tachycardia (AVRT) due to an accessory
pathway (Fig. 40-2A,B). Although heart failure does not specifically predispose to development of these two forms of PSVT (which are due to underlying abnormal electrophysiological circuits), it may increase the frequency or duration of these arrhythmias as a result of increased circulating serum catecholamine levels and electrical remodeling.
pathway (Fig. 40-2A,B). Although heart failure does not specifically predispose to development of these two forms of PSVT (which are due to underlying abnormal electrophysiological circuits), it may increase the frequency or duration of these arrhythmias as a result of increased circulating serum catecholamine levels and electrical remodeling.
Table 40-1 Supraventricular Arrhythmias | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
Treatment of PSVT requires not only that the arrhythmia be documented but also that the underlying electro-physiological mechanism be determined if possible (i.e., AVNRT or AVRT), since these two forms of PSVT may respond differently to medical treatment. The underlying mechanism of PSVT can often be delineated by electrocardiogram (ECG) if the arrhythmia can be recorded, especially if during sinus rhythm there is ventricular pre-excitation suggesting Wolff-Parkinson-White (WPW) syndrome. However, in many instances the arrhythmia is nonsustained, making ECG documentation difficult. In such a case, a 24-hour Holter monitor may record a diagnostic ECG if the arrhythmia occurs daily, or a 30-day event monitor (e.g., patient-activated or continuous-loop) may be required if the arrhythmia is less frequent. Once an ECG recording of the arrhythmia has been obtained, a determination of the underlying mechanism can be made in the majority of cases and empiric treatment begun, if indicated.
In some cases, however, documentation of the arrhythmia by ECG fails and the patient must undergo a diagnostic electrophysiology (EP) study. The EP study is a percutaneous, catheterization-based procedure, performed under light sedation, which is guided fluoroscopically or by a three-dimensional electroanatomical (contact or noncontact electrode) mapping system. To perform an EP study, typically two to four multielectrode (i.e., 4 to 20 electrodes) catheters are inserted into the heart, baseline electrical activity is recorded to identify any abnormalities in the electrical conduction system, and the heart is stimulated electrically to induce the clinical arrhythmia. During an EP study, PSVT can be induced in most patients with a history of
spontaneously occurring arrhythmias, and its underlying mechanism determined. If electrophysiological testing is ultimately required to diagnose the arrhythmia, curative ablation will usually be performed at that time (see following description).
spontaneously occurring arrhythmias, and its underlying mechanism determined. If electrophysiological testing is ultimately required to diagnose the arrhythmia, curative ablation will usually be performed at that time (see following description).
Table 40-2 Treatment of Supraventricular Arrhythmias | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
Table 40-3 Diagnosis of Supraventricular Arrhythmias | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||
The acute treatment of PSVT, as in a patient presenting to the emergency room in tachycardia, has been made simpler and safer with the development and marketing of adenosine (Adenocard). Adenosine, when given intravenously in a rapid bolus at a dose of 6 to 18 mg, will convert PSVT to sinus rhythm in the majority of patients (11,12,13). Exogenous adenosine acts by stimulating the adenosine receptor, which, mediated by a G-protein complex, activates the potassium channel IKade, causing hyperpolarization of the cell membrane and shortening of the action potential duration (14). These effects result in transient AV block that terminates the PSVT. Adenosine is very short-acting due to a half-life of 8 to 10 seconds and, consequently, causes few adverse effects. Side effects related to its use include transient flushing, chest pain, shortness of breath, and precipitation of bronchospasm in patients with a history of asthma (11,12,13,15). Adenosine may rarely cause atrial fibrillation because it shortens the atrial refractory period by activating the potassium channel IKade (16). Adenosine-induced atrial fibrillation usually terminates spontaneously in several seconds to minutes, but may (rarely) require cardioversion. In the unusual case where adenosine fails to convert PSVT, a repeat dose may be given; if an underlying WPW syndrome with pre-excitation is not suspected, intravenous verapamil 5 to 10 mg or digoxin 0.25 mg every 30 to 60 minutes up to 1 mg may be given. If WPW syndrome with pre-excitation is suspected or known to be the cause of the PSVT, then intravenous procainamide up to 100 mg per minute, as tolerated hemodynamically, up to a total dose of 1.0 g may be given. If PSVT cannot be converted pharmacologically, or in the event of hemodynamic intolerance of PSVT, direct-current cardioversion under intravenous sedation is usually a safe and very effective alternative.
Following conversion to sinus rhythm and stabilization of the patient, a decision must then be made whether longterm suppression of PSVT is required. In a patient with heart failure, it is important to prevent recurrent PSVT because of the high rate of the tachycardia (often in excess of 150 beats per minute), which may cause rapid and severe hemodynamic deterioration. In those patients with rare or infrequent episodes of PSVT, either no therapy or pharma-cologic therapy alone may be appropriate. However, in those with frequent episodes of PSVT or particularly rapid PSVT, and in those who cannot tolerate antiarrhythmic drugs, curative radiofrequency catheter ablation (RFCA) may be performed (17,18,19).
RFCA involves the endocardial delivery of radiofrequency energy (e.g., 550 kHz) via a large-tip (e.g., 4- to 10- mm) electrode catheter at the location of origin or a critical zone in the electrical circuit of the arrhythmia. Application of radiofrequency energy, at a power of 50 to 100 W achieve temperatures of 50° to 60°C, produces permanent destruction of the arrhythmogenic tissue by resistive heating.
The efficacy of RFCA is high (approaching 100% for PSVT) and major complications, including inadvertent AV block requiring pacemaker implantation, are rare, occur-ring in <1% to 3% of patients. Thus, RFCA has become first-line therapy for most patients with PSVT, particularly in view of the potential for side effects and long-term toxicity from antiarrhythmic drugs.
The efficacy of RFCA is high (approaching 100% for PSVT) and major complications, including inadvertent AV block requiring pacemaker implantation, are rare, occur-ring in <1% to 3% of patients. Thus, RFCA has become first-line therapy for most patients with PSVT, particularly in view of the potential for side effects and long-term toxicity from antiarrhythmic drugs.
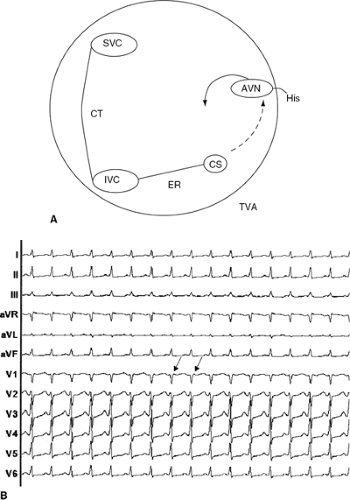 Figure 40-1 (A) A schematic diagram of the right atrium demonstrating the putative slow (dashed line) and fast (solid line) AV nodal pathways and possible re-entrant circuit of the common slow-fast form of AVNRT is shown. As noted by the gap between the head and tail of the activation wavefrant, the upper common pathway or atrial turnaround point is controversial. (B) A 12-lead ECG of typical slow-fast AV nodal re-entrant tachycardia is shown, in which antegrade AV conduction is over the slow AV nodal pathway and retrograde VA conduction is over the fast AV nodal pathway. Note the narrow QRS complex and absence of an obvious P wave, which is superimposed on the terminal portion of the QRS complex giving rise to a small terminal R’ wave seen in lead V1 (arrow). AVN, atri-oventricular node; CS, coronary sinus; His, His bundle; ER, eustachian ridge; TVA, trisucpid valve annulus. |
For patients with PSVT who are not appropriate candidates for RFCA (e.g., due to patient preference or other severe, concurrent illness), antiarrhythmic drugs may be used to prevent recurrences (Tables 40-2 and 40-3). For AVNRT or AVRT due to a concealed accessory pathway (i.e., without pre-excitation on ECG), AV nodal blocking drugs such as digoxin, calcium blockers (e.g., diltiazem or verapamil) or beta-blockers may be effective in preventing arrhythmia recurrence. However, patients with heart failure may not tolerate calcium or beta-blockers due to their negative inotropic and chronotropic effects. Furthermore, in patients with AVRT due to WPW syndrome with ventricular pre-excitation, digoxin and calcium blockers should be strictly avoided due to the potential life-threatening risk of increasing ventricular response to atrial fibrillation. By inhibiting conduction through the AV node, digoxin and calcium blockers may actually increase conduction over the accessory pathway, resulting in an irregular wide-complex ventricular response potentially in excess of 200 beats per minute, which may occasionally lead to ventricular fibrillation.
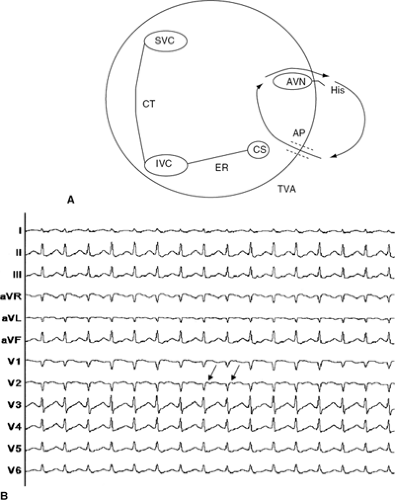 Figure 40-2 (A) A schematic diagram of the right atrium demonstrating the re-entry circuit during orthodromic AVRT is shown in a patient with a posteroseptal accessory pathway. Note the ante-grade (atrio-ventricular) conduction over the AV node and retrograde (ventriculo-atrial) conduction over the accessory pathway during orthodromic AVRT, with turnaround points in both the atrium and ventricle, (B) A 12-lead ECG of orthodromic AV re-entrant tachycardia is shown, in which antegrade AV conduction is over the AV node and retrograde VA conduction is over an accessory pathway. Note the narrow QRS complex, and the P wave visible in the early ST segment (arrow). AP, accessory pathway designated by dotted lines; AVN, atrioventricular node; CS, coronary sinus; His, His bundle; ER, eustachian ridge; TVA, trisucpid valve annulus. |
If AV nodal blocking drugs are tolerated but fail to control PSVT, the Class 1 or 3 antiarrhythmic drugs (e.g., flecainide, propafenone, ethmozine, or sotalol and amio-darone) may be effective alone or in combination with AV nodal blocking drugs. However, patients with left ventricular dysfunction and heart failure have a significantly greater risk of mortality due to life-threatening ventricular proarrhythmia from certain antiarrhythmic drugs (20,21). These arrhythmias may include torsade de pointes from Class 1a drugs such as quinidine or Class 3 drugs such as sotalol, and monomorphic ventricular tachycardia from the Class 1c drugs such as flecainide (20,21). Thus, for treatment of atrial arrhythmias in patients with heart failure the risks of antiarrhythmic drugs may outweigh their benefits (22).
If an antiarrhythmic drug is required, amiodarone may be the safest and most effective drug in patients with heart failure since it has only rarely been known to cause ventricular proarrhythmia in the form of torsade de pointes (1,2). Amiodarone, a Class 3 antiarrhythmic drug that combinel AV nodal blocking effects, sympatholytic effects, and antiarhythmic effects, is very effective in preventing recurrence of PSVT (23). Amiodarone has also been shown to have other beneficial effects in patients with heart failure (1,2). For example, amiodarone may reduce mortality in patients with nonischemic cardiomyopathy (1,2). Amiodarone may also improve ejection fraction in some patients, as well as reduce hospitalization for heart failure due to its positive inotropic effects caused by prolongation of ventricular action potential
duration (1,2). However, because of its long biological half-life (up to 30 to 90 days) amiodarone must be given initially in a loading dose of 400 to 800 mg (maximum 1,200 mg) per day for several days to 1 to 2 weeks, followed by a maintenance dose of 100 to 200 mg (maximum 400 mg) per day for rapid and continued control of most arrhythmias. The primary limitation of amiodarone is its toxic effects on specific organs, including the thyroid (hyperthyroidism or hypothyroidism), liver (elevation of transaminases), lung (interstitial pneumonitis and fibrosis), skin (hypersensitivity to sun), nervous system (painful peripheral neuropathy), and eye (corneal deposits or optic neuropathy), requiring its discontinuation in up to 20% of patients over time. Due to its long biological half-life, patients being treated with amiodarone must be evaluated for signs of organ toxicity at least every 6 months and the drug discontinued immediately at the sign of any significant toxicity, especially pulmonary toxicity, peripheral neuropathy, or hyperthyroidism.
duration (1,2). However, because of its long biological half-life (up to 30 to 90 days) amiodarone must be given initially in a loading dose of 400 to 800 mg (maximum 1,200 mg) per day for several days to 1 to 2 weeks, followed by a maintenance dose of 100 to 200 mg (maximum 400 mg) per day for rapid and continued control of most arrhythmias. The primary limitation of amiodarone is its toxic effects on specific organs, including the thyroid (hyperthyroidism or hypothyroidism), liver (elevation of transaminases), lung (interstitial pneumonitis and fibrosis), skin (hypersensitivity to sun), nervous system (painful peripheral neuropathy), and eye (corneal deposits or optic neuropathy), requiring its discontinuation in up to 20% of patients over time. Due to its long biological half-life, patients being treated with amiodarone must be evaluated for signs of organ toxicity at least every 6 months and the drug discontinued immediately at the sign of any significant toxicity, especially pulmonary toxicity, peripheral neuropathy, or hyperthyroidism.
Atrial Flutter, Re-Entrant Atrial Tachycardia, and Focal Atrial Tachycardia
Atrial flutter, re-entrant atrial tachycardia, and focal atrial tachycardia (Table 40-1) are more common than PSVT in patients with heart failure In contrast to PSVT, these supraventricular arrhythmias are confined strictly to the atrial myocardium and do not utilize the AV node or an accessory pathway as part of the arrhythmia circuit (24,25,26,27,28,29). Typical and reverse typical atrial flutter (Fig. 40-3A,B) involve reentry around the tricuspid valve annulus and are dependent on slow conduction through the cavo-tricuspid isthmus (Fig. 40-4A,B), whereas other forms of re-entrant atrial tachycardia and atypical atrial flutter involve re-entry around anatomical and/or functional obstacles such as the crista terminalis, pulmonary veins, mitral valve annulus, idiopathic or surgical scars, or patches within the atria. Due to their strictly atrial substrate, AV nodal blocking drugs (including adenosine) may slow the ventricular response but an antiarrhyth-mic drug or electrical cardioversion is usually required to convert these supraventricular arrhythmias to sinus rhythm. However, while the use of adenosine in patients with atrial flutter or atrial tachycardia is rarely therapeutic, it may be diagnostic by transiently blocking AV nodal conduction and allowing one to see the underlying atrial rhythm.
Focal atrial tachycardia differs from re-entrant atrial tachycardia in that it is due to localized automatic or triggered electrical activity that can originate from anywhere in the right or left atrium, but commonly occurs along the crista terminalis, in the coronary sinus or pulmonary veins, or along the atrioventricular valve rings. Automatic atrial tachycardia is uniquely sensitive to adenosine in some cases (30), and thus its conversion to sinus rhythm by adenosine may be both diagnostic and therapeutic. Triggered activity may result from high levels of sympathetic tone commonly seen in patients with heart failure, or from intoxication with digitalis, a drug that is commonly used in patients with heart failure (31).
For the acute pharmacologic conversion of re-entrant atnal tachycardia and atrial flutter, the recently approved antiarrhythmic drug ibutilide (1 or 2 mg over 10 to 20 minutes) has been shown to be very effective (32,36). Ibutilide is a Class 3 antiarrhythmic drug, currently available only in intravenous formulation in the United States. Ibutilide prolongs atrial and ventricular action potential duration predominately by blocking the delayed rectifier potassium channel IK, (32,33). This compound is effective in converting up to 76% of patients with atrial flutter (34,35). Its serum half-life is approximately 4 hours, during which the patient should be monitored on a hospital telemetry ward or closely in clinic (32,33,34,35). Cardiac monitoring is necessary because ibutilide prolongs the QT interval and may cause torsade de pointes ventricular tachycardia in up to 3% to 4% of patients (36). Ventricular proarrhythmia is more common with ibutilide in patients with left ventricular dysfunction (36), even though it produces no adverse hemodynamic effects (37).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


