Fig. 23.1
Juzenkai Zasshi, vol. 50, 1908, photograph courtesy of the Collections of the Department of Ophthalmology, Kanazawa University
Epidemiology
TA is an uncommon chronic inflammatory vasculitis, with an incidence of 2–3 cases per million population. It mostly young women, with a 9:1 female predominance [3, 5–7], usually Asian women [8]. In fact, TA is responsible for about 5 % of all vascular diseases in India and Japan [9].
Although TA was first described in Asia and is undoubtedly more prevalent among patients in India, Africa, and South America, this disease has a worldwide distribution, with an increasing incidence in Western countries [10].
Pathophysiology
TA most commonly involves the aortic arch and its major branches, but may involve any segment of the aorta. This condition affects the vessel walls, producing medial destruction of the aortic wall with stenosis, occlusion, or aneurysm formation. Although TA usually produces obstructive lesions, it may have a dilative component in 15 % of cases [11], and it is usually associated with disease elsewhere. However, different reports suggest a wide spectrum of prevalence, with aneurysm formation varying from 0 to 87.5 % [12, 13].
Although TA is widely known as a chronic vasculitis that affects large vessels, the intimate processes of the pathophysiology in this clinical picture remain only partially explained because they have not been completely elucidated. For this reason, TA is considered to be a multifactorial disease, with autoimmune mechanisms undoubtedly involved. In 2004, Seko et al. [14] postulated an interesting pathogenic sequence, which we will analyze in more depth. This pathologic sequence may implicate the stimulation from an antigen of unknown nature, possibly infectious, that triggers heat shock protein 65 expression in aortic tissue. This, in turn, would induce the human major histocompatibility complex class I chain-related gene A (MICA) [15].
As a response to this process, inflammatory cells, such as T-cells and natural killer (NK) cells, which express NKG2D receptors, could recognize MICA, resulting in acute inflammation. This process is increased and self-perpetuated.
Cellular Immunity
TA clearly seem to be an autoimmune disease in which cellular immunity plays a major role and humoral immunity participates in a form that still remains to be clarified. In fact, immunohistochemical studies have shown different cell strains involved in the aortoarteritis process, suggesting a critical role in the onset of the disease. Gamma-delta T cells, NK cells, cytotoxic T cells, T-helper cells, and macrophages are some of the cells found in the aortic wall. This process does not seem to start in the endothelium of the aortic wall. The pathogenesis implies the stimulation of an antigen of unknown nature. T cells, NK cells, and cytotoxic T cells express and release massive amounts perforin (a membrane-disrupting protein) through the recognition of MICA, which is directly placed on the surface of arterial vascular cells [14]. This way, the endothelial cells of the vasa vasorum are activated to allow the lymphocytes to gain access to the media and adventitia of the arterial wall [16], thus amplifying the inflammatory response, recruiting especially mononuclear cells.
Immature dendritic cells (DCs) are vastly distributed migratory cells in charge of sampling the antigens in the environment. Once activated, the profile of their chemokine receptor is modified, migrating to lymphoid tissues. Here they express a mature chemokine-producing DC profile and costimulatory molecules that interact with antigen-specific T cells.
In giant cell arteritis (also called Horton’s arteritis), DCs are activated and produce chemokines (CCL18, CCL19, and CCL21) that bind the chemokine receptor (CCR7) they express, leading to the entrapment of DC within the vascular wall [17]. The co-localization of DC near T-cells indicates that they are probably involved in the physiopathology of the TA by a similar mechanism.
Study of the cellular composition within the adventitia in Takayasu’s arteritis has shown inflammatory infiltrates containing T-cells colocalizing with dendritic cells (DC) [18 19]. This fact is essential in the development and maintenance of inflammation in the vascular wall.
The antigens (of unknown nature) are presented to killer T-cells by an epitope associated with the major histocompatibility complex (MHC) on the activated DCs, which facilitates the cytotoxic reaction present within the vascular wall.
Humoral Immunity
Although the existence of a humoral role is not known, specific immunologic markers for aortoarteritis, rheumatoid factor, and antinuclear antibodies have been identified, supporting the role of an autoimmune component [20, 21] in the establishment of the disease.
The presence of immune complexes in the sera and receptors of lymphocytes has been reported, but the results differ among authors [22].
Anti-aorta antibodies have long been reported in TA [23–28], but in the study by Baltazares [29], reactive antibodies against a total human aorta extract were searched for, including their main protein components, elastin, fibronectin, and collagen, but no difference was found compared with controls. However, in the same trial, the immunoblot technique showed an immunoprecipitate of a 45 kDa protein in the sera of TA patients significantly more frequently than in the sera of the control group [29]. In the same line of research, Eichhorn [30] suggested that an antigen to these antibodies was stored within the cytoplasm when they found that healthy cells exposed to sera from patients with TA had a bright, homogeneous, and specific immunofluorescent cytoplasm stain and weak membrane stain.
There are reports about the presence of antiphospholipid antibodies in a significant proportion of patients with TA [31–33], anticardiolipin antibodies and antimonocyte antibodies [34]. These were correlated with disease activity, suggesting a pathogenic role in TA. Finally, alpha-beta T-cells would then infiltrate the arterial wall and specifically recognize one or a few autoantigens presented by a shared epitope associated with a specific MHC on the DCs.
The participation of other proinflammatory mediators, such as interleukin (IL)-6, mainly synthesized by macrophages and T-cells, is also known. Functions of the IL-6 are to activate B-cells, enhance T-cell cytotoxicity [35] and NK cell activity [36], stimulate fibroblast proliferation, and induce acute-phase protein synthesis. Other proinflammatory molecules, such as RANTES chemokine (regulated on activation, normal T-cell-expressed and secreted) and IL-8 cytokine, are potent chemoattractants for most mononuclear cells, including those that constitute the majority of the inflammatory infiltrate of the vascular wall in TA. Interleukin-6 and RANTES can also induce the production of metalloproteinases (MMP) in the matrix from mononuclear cells and smooth muscle cells. These proteolytic enzymes degrade elastin and collagen, which are structural components of the arterial wall [37].
These findings have been reinforced by several studies in patients with TA that showed higher serum concentrations of IL-6 [38], RANTES [39], IL-8 [40], MMP-2, MMP-3, and MMP-9. The levels of these latter two MMPs are correlated with disease activity [41]. However, studies of mRNA of proinflammatory cytokines showed increased gene expression of TNF-alpha, IFN-gamma, IL-2, IL-3, and IL-4 and higher expression of IL-12 mRNA after stimulation of cells with lipopolysaccharide and lower mRNA expression of IL-10 than in controls. These results suggest a chronic establishment of the inflammatory response in Takayasu’s arteritis [42].
In conclusion, notwithstanding numerous studies, the physiopathology of TA remains unknown and is probably multifactorial, involving several actors.
The data strongly implicate of autoimmune processes, mainly led by cellular immunity, whose role remains to be elucidated. The 2004 study by Seko et al. linked some of the previously discussed data [43]. Therefore, the model proposed in Fig. 23.2 is valid for the understanding the pathogenesis of the disease. The pathologic sequence may implicate the stimulation from an unknown antigen, triggering the expression of heat shock protein 65 in the aortic wall, which in turn would induce the expression of MICA. Inflammatory cells recognize MICA and secrete perforin, disrupting the endothelial membrane of the vasa vasorum and gaining access to the arterial wall.
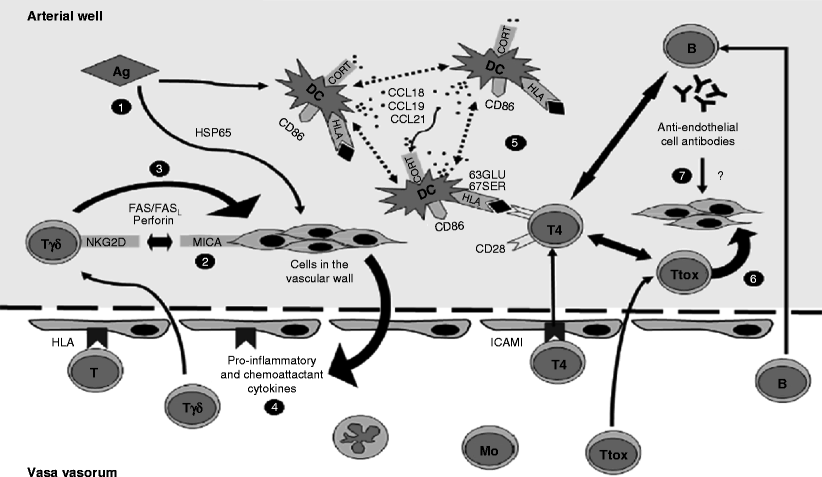
Fig. 23.2
Overview of the physiopathology of TA (brief explanation of different immunological interactions proposed by Seko’s investigations published by Arnaud in 2006) [1]. (1) Stimulation by an antigen of unknown nature triggers heat shock protein 65 expression, which in turn induces MICA. (2) Infiltrating gamma-delta T cells and NK cells expressing NKG2D receptors recognize MICA and release perforin. (3) Proinflammatory cytokines amplify the inflammatory response, inducing more MHC antigen and costimulatory molecule expression on vascular cells and thus recruiting more mononuclear cells. (4) One or a few (auto)antigens are presented to killer T-cells by a shared epitope associated with specific MHC on the activated dendritic cells trapped within the vascular wall. (5) This leads to a cytotoxic reaction of the cells within the vascular wall. (6) These dendritic cells simultaneously cooperate with B lymphocytes and induce anti-endothelial cell autoantibodies, whose role is controversial
Inflammatory mediators, such as cytokines and chemokines, are released, allowing inflammatory cells to amplify the process and entrap DCs within the arterial wall.
Alpha-beta T-cells would specifically recognize one or a few autoantigens presented by a shared epitope associated with a specific MHC on the DCs. These DC could simultaneously cooperate to some extent with the B-cells and determine a humoral immunity mainly consisting of anti-endothelial cell autoantibodies that could trigger complement-dependent cytotoxicity against endothelial cells (though this last pathogenic mechanism remains very controversial). This condition would produce medial destruction of the aortic wall with stenosis, occlusion, or aneurysm formation.
Clinical Presentation
The clinical presentation of Takayasu’s arteritis is varied and not specific. In fact, it usually develops in a very insidious way, leading the physician to consider other diagnoses, especially when the disease develops in children. Classically, this condition has two distinguishable phases, acute and chronic, and recent studies have also divided it into early (prepulseless) and late (pulseless) phases [44].
In the acute phase, the symptoms are related to the severity of the vascular inflammation. It can cause constitutional symptoms, such as weight loss, fatigue, night sweats, anorexia, malaise [45], and fever (which is more frequent in pediatric patients).
Children can present also joint pain or arthritis and extremities pain. When these symptoms are present, the differential diagnosis must be performed considering other autoimmune diseases, such as rheumatic fever and juvenile rheumatoid arthritis [46].
In the chronic phase, patients report symptoms referable to the organs involved. In a large published experience, more than half of all patients experienced upper extremity claudication, 50 % presented symptoms associated with cerebrovascular insufficiency (vision loss, lightheadedness, stroke), and a third reported carotid artery pain [13]. Less frequently, other symptoms have been reported, such as instability, vertigo, orthostatism, syncope [4], and hypertension (as a result of renal artery involvement, which was the most common presenting sign in an Indian trial) [47].
Laboratory Tests
Markers of inflammation, such as C reactive protein levels and the erythrocyte sedimentation rate, are elevated, which may help make the diagnosis, despite being very nonspecific. Multiple studies indicate that C-reactive protein levels and the erythrocyte sedimentation rate are elevated in approximately 70 % of patients in the acute phase and 50 % in the chronic phase of disease [4]. Leukocytosis and anemia may also be present, although, like the other markers, they are very nonspecific variables [48].
Imaging Study
For decades, angiography was considered the “gold standard” to confirm the diagnosis of Takayasu’s arteritis (Fig. 23.3). Although this technique can show the location and grade of the stenotic or aneurysmatic lesion [49], it is an invasive method with several limitations (radiation exposure, adverse reactions to contrast media, and arterial damage [50]). Therefore, it will probably be replaced by multiple less invasive techniques that also provide information about changes in the wall, such as ultrasonography, contrast CT scan, and MRI.
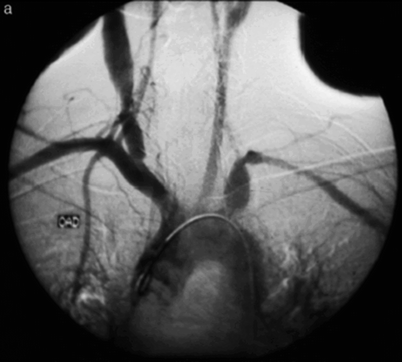
Fig. 23.3
Angiographic imaging: aneurysmal dilations of the right common artery and left intrathoracic subclavian artery
Ultrasonographic study can show the stenotic or aneurysmatic lesions and may contribute to the diagnostic process as well as to the evaluation of the grade and progress of the disease [51]. Patients with Takayasu’s arteritis can present an homogeneous, midechoic, circumferential wall thickening with luminal stenosis on carotid echo-Doppler imaging [52]. These findings have been described as pathognomonic of Takayasu’s arteritis and have been called the “macaroni sign” (Fig. 23.4) [53].
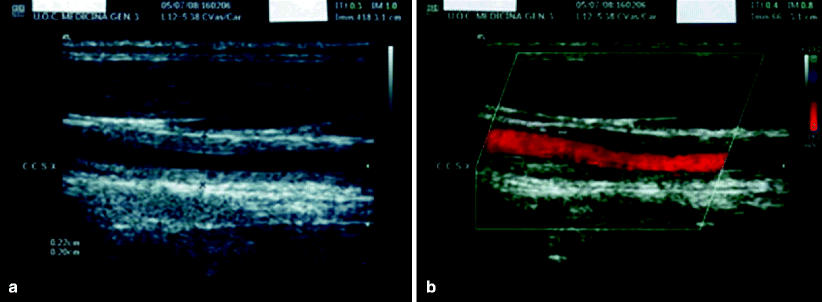
Fig. 23.4
Ultrasound B-mode (a) and color-duplex (b) flow imaging of the left common carotid artery (longitudinal section): homogeneous, midechoic, circumferential wall thickening (“macaroni sign”) with luminal stenosis. CCSX indicates the left common carotid artery. Intima-media thickness is 0.20–0.22 cm (the maximal normal value is 0.06 cm) [1]
The CT scan may have some advantages over angiography, allowing demonstration of the mural changes of the affected vessels, such as wall thickening, calcification, or thrombus, which are not seen with conventional angiography [54], but in order to determine the degree and extension of the damage to the vessels more accurately, the use of magnetic resonance imaging (MRI) may be more helpful than CT scans. MRI has other technical advantages: paramagnetic contrast media rarely cause anaphylactic reactions and are non-nephrotoxic (which can be a determinant in the pediatric patients), ionizing radiation is not used, and soft-tissue differentiation is better [55]. However, some disadvantages of MRI are that it is less available, costs more, and has more technical difficulties than CT scans.
Another diagnostic advantage of MRI compared to the CT scan is an increased sensitivity in detecting mural edema, a finding that is present in almost all acute-phase patients. However, some studies question the use of MRI as a single follow-up imaging technique because of the contradictory findings of disease activity in patients with Takayasu’s arteritis [56].
The use of 18F-FDG PET and enhanced CT has also been described to determine the inflammatory status of the vessels. Parallel studies show that these tests can show the distribution of the inflammation in the aorta, its branches, and the pulmonary artery, suggesting that the 18F-FDG accumulation observed in Takayasu’s arteritis patients directly indicates the inflammation in the vascular wall [57].
Diagnostic Criteria and Classification
Several diagnostic criteria models have been proposed in the literature [58, 59]. In 1988, Ishikawa [60] published his clinical criteria to establish the diagnosis of Takayasu’s arteritis. These proposed criteria were based on clinical and angiographic data from 96 patients with Takayasu’s disease.
One obligatory criterion: age ≤40 years.
Two major criteria: left and right mid-subclavian artery lesions.
Nine minor criteria: high erythrocyte sedimentation rate, common carotid artery tenderness, hypertension, aortic regurgitation, annuloaortic ectasia, lesions of the pulmonary artery, left mid common carotid artery, distal brachiocephalic trunk, thoracic aorta, and abdominal aorta.
In addition to the obligatory criteria, the presence of two major criteria, one major plus two or more minor criteria, or four or more minor criteria suggests a high probability of the presence of Takayasu’s disease. The criteria had 84 % sensitivity in 96 patients with this disease.
In 1990, The American College of Rheumatology [61] established the current and most widely used diagnostic criteria. They proposed that three or more criteria must be present to make the diagnosis:
1.
Age of onset of illness 40 years or less. Development of symptoms or findings related to Takayasu’s arteritis at age 40 or less.
2.
Claudication of extremities. Development and worsening of fatigue and discomfort in the muscles of one or more limbs with activity, especially the upper extremities.
3.
Decreased brachial artery pulse. Decreased pulsation of one or both brachial arteries.
4.
Differential blood pressure greater than 10 mmHg. Differential pressure greater than 10 mmHg systolic between arms.
5.
Murmur over subclavian arteries or aorta. Audible murmur on auscultation on one or both subclavian arteries or abdominal aorta.
6.
Abnormal arteriography. Arteriographic narrowing or occlusion of all primary aortic branches or large arteries in the proximal upper and lower extremities not due to arteriosclerosis, fibromuscular dysplasia, or similar causes: changes usually focal or segmental.
Three or more criteria provide a sensitivity and specificity for diagnosis of 90.5 % and 97.8 %, respectively [13].
Concerning the classification of TA, in 1996 Hata et al. [62] published the classification currently used, which grouped the different presentations into five types depending on the affected segments, adding two subtypes depending on the involvement of the pulmonary or coronary artery.
In 2004, Nastri [63] published a diagram of this new classification.
Type I. Aortic arch vessels
Type IIA. Ascending Aorta, aortic arch, and branches
Type IIB. Type IIA vessels plus descending aorta
Type III. Descending and abdominal aorta and/or renal artery
Type IV. Abdominal aorta and renal artery
Type V. Combination of types IIB and IV (Fig. 23.5)
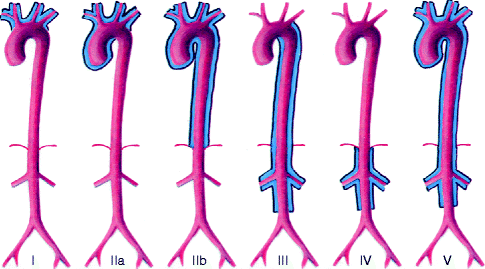
Fig. 23.5
Classification of Takayasu’s arteritis according to the anatomic site of involvement
Aneurysmal Disease of Takayasu’s Arteritis
The specific incidence of aneurysmal presentation is not known, but it is more frequent in Japan, India, Thailand, Mexico, and Africa [64, 65], compared with occlusive disease, which is more prevalent in the United States and Europe. It is not quite clear why there is such a difference. Frequently, aneurysm formation develops in the descending aorta, followed by the abdominal aorta, and, less frequently, the ascending aortic segments.
Aneurysm formation is considered one of the main complications in Takayasu’s arteritis and is an extreme marker of disease activity, usually present in patients with a long history of disease, implying a poor prognosis. Miyata published a comparison of survival rates of patients with Takayasu’s arteritis based on the presence or absence of aneurysms, showing a 66.5 % survival rate in patients with aneurysms versus a 79.5 % survival rate in patients without them [66] in 15 years of follow-up.
Although the incidence of aneurysmal presentation of Takayasu’s disease is unknown, some authors have provided information on small groups of patients. These studies show a moderately high incidence of aneurysms.
Matsumura in 1991 showed an incidence of saccular or fusiform aneurysms in 31.9 % (36/113) of patients with Takayasu’s arteritis. Multiple aneurysms were found in 15 patients. In their study, most patients were over 40 years old, and most of the aneurisms were located in the ascending aorta [65]. In 1994, Kerr et al. [45] published a series of 60 patients with Takayasu’s arteritis from the National Institute of Health. Twenty-three percent had aortic aneurysm formation. In 2000, Sueyoshi, from Omura Hospital, showed 17 aortic aneurysms in 14 (45.2 %) of 31 patients [67].
It’ is curious that Nakao [25], in his series of 84 patients, revealed no cases of aneurysmal dilatation of the aorta, describing only 6 cases of moderate post-stenotic dilatation in the angiographic findings. In Chile, Krämer reported that the incidence of aneurysm formation was less than 10 % [68].
Thoracic and Abdominal Aortic Aneurysms
Thoracic and abdominal aneurysms are the most common aneurysm presentations in Takayasu’s arteritis [69]. Matsumura showed a markedly high frequency in the thoracic area (29 thoracic and 7 abdominal, respectively) [65]. However, Sueyoshi [13] showed a proportional distribution between the thorax and abdomen (9 and 8 patients, respectively). In 2004, Kieffer et al. in Paris [70] published a series of 27 years’ experience, including 10 thoracic and 23 thoracoabdominal aneurysms. Six patients had associated aneurysms of the proximal aorta.
Extracranial Supraaortic Aneurysm
Aneurysms also occur in the subclavian [71], innominate, and carotid arteries (Regina et al., 1998 [72]), but extracranial carotid aneurysms caused by Takayasu’s arteritis are extremely rare [73]. Tabata et al. [74] reported six cases of extracranial carotid aneurysm among 106 cases in 50 years.
These anatomical presentations may have surgical difficulties in the resection and replacement of the arteries, with a relatively high incidence of stroke. Therefore, decision-making in these cases must be tailored to the individual, depending upon the extent of involvement and the symptoms, and also evaluating the possibility of hybrid intervention with the use of endovascular stent grafts [75] (Fig. 23.6).
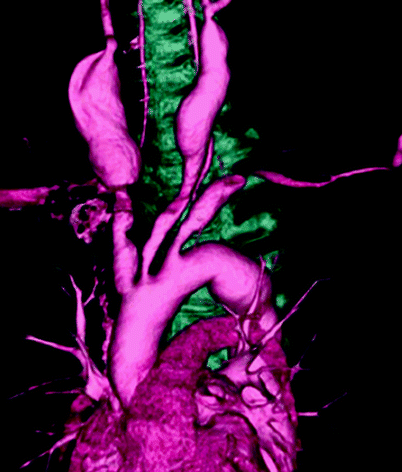
Fig 23.6
Bilateral common carotid artery aneurysm in Takayasu’’s arteritis: Tochii M. Asian Cardiovasc Thorac Ann. 2008;16:185–186
Coronary Lesions
Coronary artery involvement in Takayasu’s arteritis was first described by Frovig in 1951, and the first coronary artery bypass grafting was performed by Young [76] in 1973.
Endo et al. [3] in 2003 published their experience in coronary angiography in 130 patients with Takayasu’s arteritis. Thirty-one had coronary artery lesions, and only four had aneurysmal coronary ectasia (Fig. 23.7). In their experience, aneurysms were not treated surgically because they were diffuse and the patients had no angina.
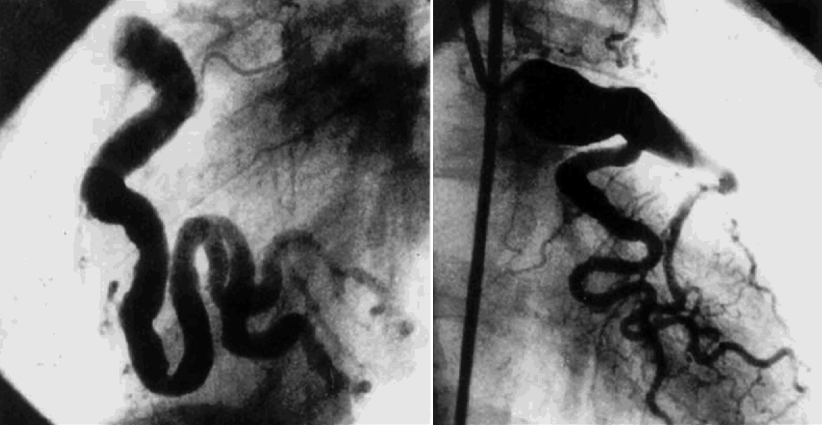
Fig. 23.7
Diffuse dilatation of the right and left coronaries arteries in a patient with Takayasu’s arteritis. Endo M: J Tho Card Surg 2003;125(3):570–7
The incidence of coronary lesions complicating Takayasu’s arteritis is relatively low. Nasu [77] in 1976 reported coronary lesions complicating Takayasu’s arteritis in 10.5 % (8/76 autopsy cases). More recently, Amano and Zuzuki [78] reported 45 % of coronary lesions, of which 73 % are occlusive coronary lesions localized around the coronary ostia as a result of the extension of inflammation-induced intimal proliferation and fibrous contraction from the ascending aorta.
Treatment and Management
As mentioned earlier, Takayasu’s arteritis is a chronic disease, which in its initial phase has only nonspecific symptoms, making the differential diagnosis difficult. This is the challenge for the clinician, because in the chronic phase, the symptoms are actually signs of a long process of complications, such as stenosis or aneurysmal formations.
According to the study published by Koide [79] about the therapeutic approach in Japan in the 1980s, the vast majority of patients received drug therapy alone (75 %), and surgery was a viable option in 12 % of patients. With the current technical advances, especially the development of different endovascular techniques, surgery has become a more popular therapy [80].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


