Lymphedema
Stanley Rockson
Steven M. Dean
Lymphedema is the broad descriptive term that is employed to describe the clinical presentation of lymphatic vascular insufficiency. In lymphatic disease, there is excessive, regionalized (typically) interstitial accumulation of protein-rich fluid. Such conditions develop when there is an imbalance between the lymphatic load (the amount of lymph generated at the tissue level) and the lymphatic transport capacity; either or both of these variables may contribute to the development of clinically relevant lymphatic edema. Lymphedema can occur as a consequence of heritable disorders of lymphatic vascular development (primary) or as an acquired condition (secondary); the latter typically results as a consequence of traumatic disruption of the structural or functional integrity of the lymphatic circulation. Although lymphedema most typically occurs as a consequence of abnormal regional lymphatic drainage of one or more of the extremities, visceral involvement, particularly in the respiratory and gastrointestinal tracts, is occasionally encountered. Clinically, lymphedema of the extremities will produce chronic volume excess in the affected limbs; with time, irreversible structural changes supervene in the skin and soft tissues of the affected regions. In addition, lymphedema produces a well-documented, detrimental effect on the patient’s perceived well-being and quality of life. The associated psychological derangements may, in turn, reduce compliance with recommended therapeutic interventions.
CLINICAL FEATURES
Clinical Classification
The clinical evaluation of lymphedema entails the differentiation of so-called primary and secondary causes (Table 26.1).
Primary lymphedema may be present at birth or develop at predictable points in the patient’s natural history. Thus, the primary lymphedemas are typically classified according to the age of onset of edema. Congenital lymphedema is detectable at birth or within the first 2 years of life. Lymphedema praecox begins at, or shortly after, the onset of puberty but may be delayed for up to two decades. Lymphedema tarda most commonly appears after age 35 but is relatively uncommon, accounting for less than 10% of cases. Many of the primary lymphedemas are inherited in an autosomal dominant pattern, although there is, despite the Mendelian form of inheritance, a modest-to-significant female predominance in the clinical presentation. The most common form of congenital lymphedema carries the eponym of Milroy’s disease, which is typically bilateral in distribution. The molecular basis of this disease has been established in numerous family
cohorts: the disease appears to be the reflection of disordered lymphatic vasculogenesis, since it is determined by the presence of missense mutations in the domain encoding the tyrosine kinase signaling function of the vascular endothelial growth factor receptor 3 (VEGFR3) and the lymphatic endothelial receptor that mediates the effects of VEGF-C and VEGF-D. Other genetically determined syndromes include lymphedema-distichiasis, which has been ascribed to mutations in the nuclear transcription factor FOXC2, and cholestasis-lymphedema syndrome. Lymphedema praecox is the most common form of primary lymphedema, accounting for up to 94% of the cases in large reported series. Here, there is an estimated 10:1 ratio of females to males. The edema is most often unilateral and limited to the foot and calf.
cohorts: the disease appears to be the reflection of disordered lymphatic vasculogenesis, since it is determined by the presence of missense mutations in the domain encoding the tyrosine kinase signaling function of the vascular endothelial growth factor receptor 3 (VEGFR3) and the lymphatic endothelial receptor that mediates the effects of VEGF-C and VEGF-D. Other genetically determined syndromes include lymphedema-distichiasis, which has been ascribed to mutations in the nuclear transcription factor FOXC2, and cholestasis-lymphedema syndrome. Lymphedema praecox is the most common form of primary lymphedema, accounting for up to 94% of the cases in large reported series. Here, there is an estimated 10:1 ratio of females to males. The edema is most often unilateral and limited to the foot and calf.
TABLE 26.1 CLASSIFICATION SCHEMA FOR LYMPHEDEMA | ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||
Secondary lymphedemas are much more common than the primary disorders and develop when the lymphatic vascular channels or nodal structures are damaged or obstructed by other processes, most commonly due to surgery, infection, trauma, or radiotherapy. Figure 26.1 demonstrates a case of secondary lymphedema due to axillary lymph node dissection (following breast cancer). In the United States, lymphedema following axillary lymph node dissection is the most common cause of secondary lymphedema (˜15% in breast cancer patients). Similar edematous complications in the lower extremities occur following interventions for pelvic and genital cancers, particularly when inguinal and pelvic lymph nodes are dissected or irradiated. Lymphedema is a common sequel after lymph node dissection for malignant melanoma as well. Worldwide, the most common cause of secondary lymphedema is filariasis, where severe forms of lymphatic destruction can lead to the advanced presentations of lymphedema, called elephantiasis. Morbid obesity is an increasingly prevalent, yet underappreciated, secondary cause of lower-extremity lymphedema (Fig. 26.2).
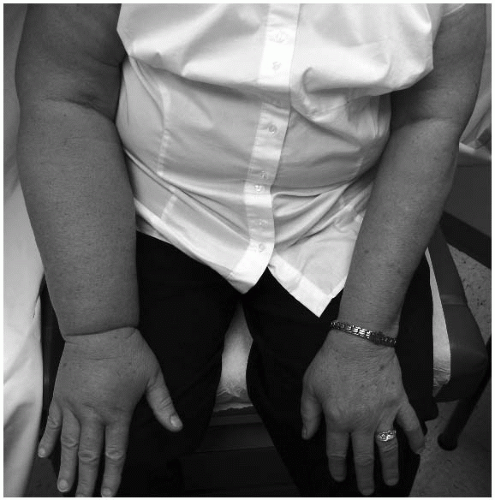 FIGURE 26-1. Secondary lymphedema of the right upper extremity from a prior axillary lymph node dissection for breast carcinoma. |
Physiology and Pathophysiology
The lymphatic vasculature begins, in the skin, in the form of blind-end sinuses, called the initial lymphatics. Attached to the surrounding connective tissue by anchoring filaments, and characterized by the absence of tight intercellular junctions, the initial lymphatics are ideally designed to encourage the rapid ingress of extracellular fluid, macromolecules, and cells through the porous basement membrane. The initial lymphatics ultimately coalesce into lymphatic conduit vessels that are invested with unidirectional valvular structures, resembling those of the venous circulation, and a prominent smooth muscle layer that possesses remarkable autocontractile properties. Lymph flow ultimately depends on lymphatic vascular contraction that can, in turn, be influenced by neural and humoral factors. Lymph flow and contractility increase in the presence of increased tissue turgor, upright posture (hydrostatic forces), mechanical stimulation, and exercise. The conduit vessels carry lymph into the lymph nodes, where lymphovenous fluid exchange is able to occur. In the extremities, the superficial (epifascial)
system collects lymph from the skin and subcutaneous tissues; the deep system carries the lymph that originates in the subfascial structures (muscle, bone, and blood vessels). In the lower extremity, the superficial and deep systems merge within the pelvis, whereas those of the upper extremity coalesce in the axilla. Functional or structural disruption of any component of this intricate drainage system can lead to the accumulation of proteinrich fluid in the interstitium. A variety of anatomic and functional derangements can lead to lymph stasis. Mutations identified in genes that regulate lymphatic development can result in inherited lymphedema. Recent studies in families with inherited forms of lymphedema have identified the following six genes that cause lymphedema: FLT 4 (encoding VEGFR3), FOXC2, SOX18, HGF/MET, HGF, GJC2, and CCBE1. Anatomically, these gene mutations may manifest as lymphatic hypoplasia, functional insufficiency, or anatomic absence of lymphatic valves, leading to lymphangiectasia, or a hypothesized intrinsic derangement in the contractile function of the vasculature. Secondary lymphedema, which is much more common than the primary form, can appear as a by-product of any trauma, inflammation, radiation, or surgical disruption of the lymphatic vascular or nodal structures. Cellulitis may actually be more of a predictor of underlying, initially subclinical, lymphedema as opposed to a cause of the subsequent clinically manifest lymphedema. The significant role of obesity in provoking
secondary lymphedema cannot be overemphasized. It must be recognized that patients with “occult” or asymptomatic primary lymphedema are at risk for developing clinical lymphedema when exposed to a “secondary” insult. Primary involvement of the lymphatics with neoplasia is a common setting for the development of secondary lymphedema.
system collects lymph from the skin and subcutaneous tissues; the deep system carries the lymph that originates in the subfascial structures (muscle, bone, and blood vessels). In the lower extremity, the superficial and deep systems merge within the pelvis, whereas those of the upper extremity coalesce in the axilla. Functional or structural disruption of any component of this intricate drainage system can lead to the accumulation of proteinrich fluid in the interstitium. A variety of anatomic and functional derangements can lead to lymph stasis. Mutations identified in genes that regulate lymphatic development can result in inherited lymphedema. Recent studies in families with inherited forms of lymphedema have identified the following six genes that cause lymphedema: FLT 4 (encoding VEGFR3), FOXC2, SOX18, HGF/MET, HGF, GJC2, and CCBE1. Anatomically, these gene mutations may manifest as lymphatic hypoplasia, functional insufficiency, or anatomic absence of lymphatic valves, leading to lymphangiectasia, or a hypothesized intrinsic derangement in the contractile function of the vasculature. Secondary lymphedema, which is much more common than the primary form, can appear as a by-product of any trauma, inflammation, radiation, or surgical disruption of the lymphatic vascular or nodal structures. Cellulitis may actually be more of a predictor of underlying, initially subclinical, lymphedema as opposed to a cause of the subsequent clinically manifest lymphedema. The significant role of obesity in provoking
secondary lymphedema cannot be overemphasized. It must be recognized that patients with “occult” or asymptomatic primary lymphedema are at risk for developing clinical lymphedema when exposed to a “secondary” insult. Primary involvement of the lymphatics with neoplasia is a common setting for the development of secondary lymphedema.
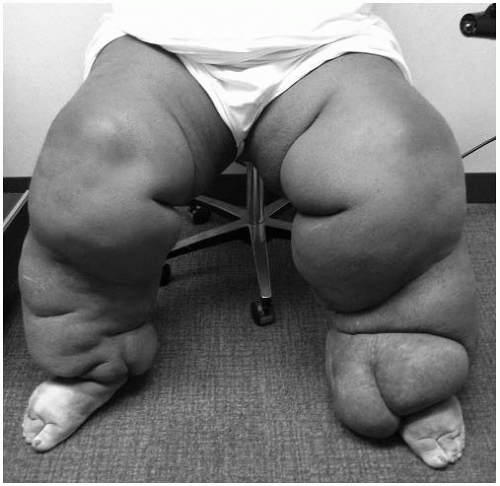 FIGURE 26-2. Morbid obesity is an increasingly prevalent, yet underappreciated, secondary cause of lower-extremity lymphedema. |
The commonly accepted concepts of lymphedema pathophysiology are derived from observations in experimental animal model systems. When mechanical interruption of lymph vessels is created in the canine limb, lymphedema arises after an elapsed latency phase of months to years. Prior to the appearance of overt edema or any measurable accumulation of interstitial fluid, there is demonstrable anatomic pathology if the conduit lymphatic vessels are angiographically imaged. Scanning electron microscopy demonstrates irreversible loss of competence of the interendothelial junctions. Eventually, the lymph collectors become fibrotic and permeability is lost. In contrast to what is seen in hydrostatic edema, however, chronic lymphedema is not characterized by lymphatic hypertension.
Chronic lymph stasis fosters the extracellular accumulation of protein and cellular metabolites, including hyaluronan and other glycosaminoglycans. The increased tissue colloid osmotic pressure leads to water accumulation and elevation of interstitial hydraulic pressure. Changes in the tissue architecture are common. Lymph stagnation is often accompanied by an increase in the presence of fibroblasts, adipocytes, and keratinocytes in the affected regions. Enhanced populations of mononuclear cells (chiefly macrophages) signal the presence of a chronic inflammatory response. There is an exaggerated deposition of collagen, ultimately accompanied by adipose and connective tissue overgrowth in the edematous skin and subcutaneous tissues. The histopathology of chronic lymphedema may include thickening of the lymphatic basement membranes, fragmentation and degeneration of elastin, prominent ingress of fibroblasts and inflammatory cells, and increased amounts of ground substance and pathological collagen fibers. Clinically, these histologic features contribute to the development of progressive cutaneous and subcutaneous fibrosis. Table 26.2 summarizes the pathophysiology of lymphedema.
TABLE 26.2 PATHOPHYSIOLOGY OF LYMPHEDEMA | ||||||||
|---|---|---|---|---|---|---|---|---|
|
Presentation and Common Clinical Signs and Symptoms
Natural History of Lymphedema
The natural history of lymphedema can be quite variable. A large percentage of the patients with objectively documented lymphatic dysfunction remain asymptomatic. A long phase of latency characterizes both primary and secondary forms of lymphedema. The latent phase terminates when compensatory mechanisms become inadequate to withstand the requirement for lymphatic flow.
The precipitating factors for the transformation to overt lymphedema are not known. Initially, mild increases in limb volume may be intermittent and may, in fact, elude the patient’s notice. Nevertheless, in most patients, the maximal increase in girth of the involved limb is determined within the first year after onset, unless there are supervening complications such as recurrent cellulitis. The propensity to recurrent soft tissue infection is one of the most troublesome aspects of long-standing lymphedema. Bacterial growth is encouraged both by the boggy, protein-enriched nature of the interstitium and by the impaired regional immune responses that reflect the abnormal immune traffic of lymphedema. Recurrent attacks of cellulitis further damage the cutaneous lymphatics, worsen the skin quality, and aggravate the edema.
Signs and Symptoms of Lymphedema
The clinical stages of lymphedema are outlined in Table 26.3




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
