Treatment algorithms in pediatric pulmonary arterial hypertension (PAH) are derived from clinical trials in adult populations and from clinical practice, but experience in children is limited. In this retrospective cohort study, we analyzed outcomes in a previously identified cohort of 86 consecutive children with PAH treated with bosentan as part of their treatment regimen. All children with idiopathic PAH or heritable PAH and PAH associated with congenital heart disease or connective tissue disease who started bosentan treatment from May 2001 to April 2003 in 2 tertiary pediatric referral centers were followed, with data collection ending August 2006. Eighty-six children (37 male, 49 female) 11 ± 5 years of age with idiopathic/heritable PAH (n = 36), PAH associated with congenital heart disease (n = 48), or PAH associated with connective tissue disease (n = 2) received bosentan as monotherapy (n = 42) or as an add-on to pre-existing continuous intravenous epoprostenol or subcutaneous treprostinil (n = 44). Median observation period was 39 months (range 2 to 60). Thirty-four patients (40%) received ≥1 additional PAH-specific therapy during follow-up. At end of data collection, 25 patients (29%) remained on bosentan, 43 (50%) had stopped bosentan, 11 (13%) had died while on bosentan, and 7 were lost to follow-up. At 4 years, the Kaplan-Meier estimate of disease progression in patients while on bosentan was 54% (7 patients at risk) with a survival estimate of 82% (16 patients at risk). Risk factors significantly associated with survival were World Health Organization functional class and indexed pulmonary vascular resistance. In conclusion, outcome in children with PAH managed with current treatment regimens appears favorable. However, despite current therapy options, disease progression remains a concern.
Pulmonary arterial hypertension (PAH) is a progressive disease characterized by remodeling of the pulmonary vasculature resulting in right ventricular failure and death, if untreated. In children, PAH is most often idiopathic (IPAH), heritable (HPAH), or associated with congenital heart disease (PAH-CHD). Bosentan is an orally active dual endothelin receptor antagonist that delays time to clinical worsening and improves exercise capacity and hemodynamics in adult patients with PAH. In children, the effectiveness and safety of bosentan is supported by prospective and retrospective studies. However, long-term outcome data remain limited. In this retrospective cohort study, we describe long-term disease progression and survival in a previously identified cohort of 86 consecutive children with PAH in whom treatment with bosentan was initiated as part of their treatment regimens from May 2001 to April 2003 until end of data collection, August 2006. These data provide real-world experience with such treatment regimens in the management of children with PAH.
Methods
This is a retrospective cohort study in 86 consecutive pediatric patients with PAH (≤18 years of age at bosentan initiation) who started bosentan from May 2001 to April 2003 with or without pre-existing continuous parenteral intravenous epoprostenol or subcutaneous treprostinil therapy at 2 tertiary referral centers in the United States (Columbia University Medical Center, New York, New York, and the Children’s Hospital, Denver, Colorado). Follow-up data were collected retrospectively from medical charts; end of data collection was August 31, 2006. Patient characteristics and treatment algorithms followed in the 2 centers have been described previously. PAH was confirmed by right heart catheterization. This study was approved by the respective institutional review boards.
Bosentan treatment was initiated as monotherapy or as an add-on to pre-existing therapy with intravenous epoprostenol or subcutaneous treprostinil. The maintenance dose of bosentan, the dose of pre-existing medications (intravenous epoprostenol, subcutaneous treprostinil), and additional PAH-specific therapies initiated during the observation period were adjusted according to the treating physicians’ clinical judgment. Reasons for addition of PAH-specific therapies were “further improvement desired,” “deterioration,” or “other.” Cardiopulmonary hemodynamic parameters (cardiac index, mean pulmonary arterial pressure, pulmonary vascular resistance index, and mean right atrial pressure) were obtained by cardiac catheterization (children were anesthetized) within the 3-month period preceding bosentan initiation. World Health Organization (WHO) functional class (FC) and 6-minute walk distance (6MWD; if age appropriate) were assessed before starting bosentan treatment and during follow-up. Addition of PAH-specific therapy was recorded. The composite end point “disease progression” was predefined as the first occurrence of ≥1 of the following events: death, atrial septostomy, lung transplantation, deterioration in WHO FC compared to baseline, decrease of ≥15% in 6MWD compared to baseline, discontinuation of bosentan due to PAH deterioration according to the treating physician’s clinical judgment, and/or addition of PAH-specific therapy due to PAH deterioration after the initiation of bosentan. Survival status and treatment were assessed for all patients at end of data collection, regardless of whether bosentan was continued or discontinued. Peripheral edema, systemic hypotension, increases in hepatic transaminase levels, and events leading to discontinuation were collected as safety experience. Due to the small number of patients with PAH associated with connective tissue disease (n = 2), only demographics are presented for this subgroup.
Due to the retrospective nature of the data collection, statistical analysis was descriptive and exploratory. Person-time incidence rates were calculated by dividing the number of events by the cumulative treatment exposure. The p values are not reported because there was no predefined study hypothesis, reflecting Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidance. Proportions were reported with 95% 2-sided confidence intervals (CIs) based on the binomial exact distribution. Patients who died were assigned WHO FC IV. Time to addition of PAH-specific therapy for any reason, time to disease progression, and time to death were summarized using Kaplan-Meier estimates and 95% CIs (patients lost to follow-up were censored).
Univariate Cox regression analyses for survival were performed and risk factors significant at the 0.10 level were noted. However, multivariate Cox proportional-hazards regression analysis was based on a backward selection process with a selection level of 0.0157. Prespecified risk factors at bosentan initiation were sex, age at PAH diagnosis, time from PAH diagnosis to initiation of bosentan, cause (e.g., IPAH/HPAH vs PAH other), WHO FC (i.e., WHO FC I/II vs III/IV), and monotherapy at bosentan initiation (i.e., bosentan without background prostanoid therapy). Hemodynamic variables (cardiac index, mean pulmonary arterial pressure, pulmonary vascular resistance index, and mean right atrial pressure) were also included in an additional Cox regression analysis despite a large proportion of missing data. Each continuous risk factor was transformed into a binary variable using its mean as a cutoff.
Results
In this cohort of 86 children with PAH, the median observation period, defined as time between initiation of bosentan and date of last clinical information capture before end of data collection, was 39 months (mean ± SD 35 ± 15, range 2 to 60).
Demographic characteristics at bosentan initiation have been presented previously ( Table 1 ). Patients’ ages ranged from 9 months to 18 years at the start of bosentan therapy. More patients with PAH-CHD were female and started bosentan as monotherapy. Congenital heart defects were fully repaired in 19 patients (40%, including 4 patients on pre-existing prostanoid therapy) or partially repaired or unrepaired for 29 patients (60%, including 14 patients on pre-existing prostanoid therapy). A larger percentage of patients on pre-existing prostanoid therapy had partially repaired or unrepaired congenital heart defects compared to repaired defects at bosentan initiation than patients not on prostanoid therapy (78% vs 50%). Hemodynamic parameters were also consistent with more severe disease in the subgroup with pre-existing prostanoid therapy at bosentan initiation ( Table 1 ). Congenital heart defects included (repaired/partially repaired and unrepaired) atrial septal defects (1/10), ventricular septal defects (5/9), patent ductus arteriosus (2/5), atrioventricular septal defects (3/0), transposition of the great arteries (3/1), tetralogy of Fallot (3/1), “absent” isolated pulmonary artery (1/1), single ventricle (0/1), total anomalous pulmonary venous return (1/0), and interrupted aortic arch with ventricular septal defect (0/1). Nineteen patients had Eisenmenger syndrome, defined as unrestrictive and unrepaired CHD with systemic arterial oxygen saturation in room air <90% at rest. Median 6MWD was 456 m (mean ± SD 473 ± 105, range 201 to 719) in 53 patients who were old enough to perform the 6MWD.
| Treatment at bosentan initiation (number of patients) | All Patients ⁎ (n = 86) | Patients With IPAH/HPAH (n = 36) | Patients With PAH-CHD (n = 48) | ||
|---|---|---|---|---|---|
| monotherapy (11) | plus prostanoid (25) | monotherapy (30) | plus prostanoid (18) | ||
| Male/female | 37/49 | 6/5 | 14/11 | 9/21 | 6/12 |
| Age | |||||
| At bosentan initiation | 11 ± 5 (0–18) | 10 ± 6 (1–16) | 11 ± 4 (4–16) | 10 ± 6 (0–18) | 12 ± 4 (6–18) |
| At pulmonary arterial hypertension diagnosis | 5 ± 5 (0–16) | 8 ± 5 (1–15) | 5 ± 5 (0–16) | 3 ± 4 (0–13) | 6 ± 4 (1–15) |
| Weight at bosentan initiation | 34 ± 16 (5–89) | 35 ± 17 (8–62) | 37 ± 17 (14–89) | 30 ± 16 (5–64) | 37 ± 15 (17–57) |
| Hemodynamic parameters † | |||||
| Mean pulmonary artery pressure (mm Hg) | 63 ± 20 (67) | 58 ± 18 (10) | 65 ± 26 (17) | 60 ± 16 (26) | 75 ± 20 (12) |
| Mean pulmonary capillary wedge pressure (mm Hg) | 9 ± 3 (67) | 9 ± 2 (10) | 8 ± 3 (17) | 9 ± 4 (26) | 9 ± 3 (12) |
| Mean right atrial pressure (mm Hg) | 7 ± 4 (67) | 5 ± 2 (10) | 7 ± 4 (17) | 7 ± 4 (26) | 9 ± 3 (12) |
| Cardiac index (L/min/m 2 ) | 3.6 ± 1.4 (62) | 3.7 ± 1.3 (10) | 3.8 ± 1.2 (17) | 3.3 ± 1.3 (23) | 4.4 ± 2.0 (10) |
| Pulmonary vascular resistance index (Woods units × m 2 ) | 20 ± 14 (66) | 16 ± 13 (10) | 20 ± 17 (17) | 22 ± 14 (25) | 21 ± 12 (12) |
| World Heath Organization functional class I/II/III/IV | 6/36/33/6 | 0/4/6/1 | 3/8/10/2 | 2/16/9/2 | 1/8/7/0 |
⁎ Two male patients with PAH associated with connective tissue disease were included in the all-patient group: 1 patient had bosentan monotherapy and 1 had pre-existing prostanoid treatment at baseline; they were 8 and 10 years old at initiation and at diagnosis, they weighed 22 and 43 kg, and their mean pulmonary arterial pressures were 38 and 54 mm Hg, respectively.
† Most recent right heart catheterization within 3 months of bosentan initiation.
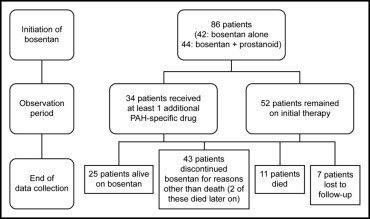
Median bosentan exposure time up to end of data collection was 24 months (mean ± SD 29 ± 17, range 2 to 63). Treatment patterns of patients throughout the observation period are presented in Figure 1 . Thirty-four of the 86 patients (40%) had ≥1 additional PAH-specific therapy added during the follow-up period; phosphodiesterase Type 5 (PDE-5) inhibitors were added in 30 of these 34 patients (88%) and prostanoids were added in 15 patients (44%). The most frequent reason for adding PDE-5 inhibitors was further improvement desired (n = 24); for adding prostanoids it was deterioration (n = 11). Kaplan-Meier estimates of time to addition of PAH-specific therapy at 1 year and 2, 3, and 4 years are listed in Table 2 . Overall, dose of epoprostenol for the 36 children treated with pre-existing epoprostenol at bosentan initiation decreased from 73 ± 42 (mean ± SD) to 46 ± 45 ng/kg/min at end of data collection; the dose for the 8 patients treated with subcutaneous treprostinil at bosentan initiation decreased from 59 ± 26 (mean ± SD) to 50 ± 47 ng/kg/min.
| Year | Time to Addition of PAH-Specific Therapy | Patients Remaining at Risk of Event | Time to Disease Progression | Patients Remaining at Risk of Event |
|---|---|---|---|---|
| 1 | 8% (95% CI 2–13) | 64 | 21% (95% CI 12–29) | 60 |
| 2 | 36% (95% CI 24–47) | 26 | 47% (95% CI 35–58) | 32 |
| 3 | 62% (95% CI 47–78) | 10 | 56% (95% CI 44–68) | 20 |
| 4 | 70% (95% CI 54–86) | 4 | 67% (95% CI 54–80) | 6 |
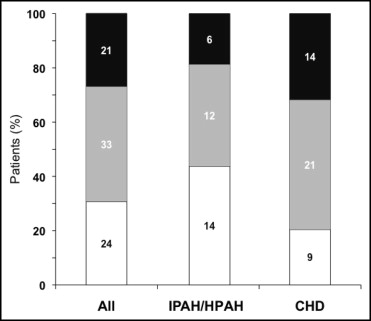
Seventy-eight of the 86 patients who had WHO FC data at bosentan initiation and ≥1 assessment during the observation period were evaluated for change in WHO FC. Median exposure to bosentan in these patients was 24 months (mean ± SD 29 ± 17, range 2 to 63). Percentages of patients with improved and worsened WHO FC at the last assessment are displayed in Figure 2 . At this time point, 24 patients (31%) showed improvement (95% CI 21 to 42), and 21 patients (27%) showed worsening (95% CI 18 to 38). WHO FC improved in 44% of patients with IPAH/HPAH and in 20% of patients with PAH-CHD.
At end of data collection, 25 of the 86 patients (29%) remained alive on bosentan ( Figure 1 , Table 3 ). All 25 patients received ≥1 additional specific (prostanoid, n = 18, or PDE-5 inhibitor, n = 18) or nonspecific (warfarin, n = 18; oxygen, n = 14; diuretics, n = 12; calcium channel blocker, n = 5; dipyridamole, n = 3; L-arginine n = 2) PAH treatment or underwent a palliative atrial septostomy (n = 1). Forty-three of the 86 patients (50%) discontinued bosentan before end of data collection owing to lack of adequate improvement based on parents’/patient’s or physician’s perception (n = 19), PAH deterioration (n = 9), increases in liver enzymes (n = 5), other adverse events (n = 5), other reasons (n = 4), or transplantation (n = 1). Two of the 43 patients who discontinued bosentan before end of data collection died 11 and 34 months after bosentan discontinuation. Twenty-nine of the 43 patients (67%) who discontinued bosentan before end of data collection were treated with up to 3 subsequent or concomitant PAH-specific treatments including prostanoids (n = 19), PDE-5 inhibitors (n = 17), and endothelin receptor antagonists (n = 12, including 1 patient in whom bosentan was reintroduced). Of the 86 patients, 11 (13%) died while on bosentan. Out of these, 5 patients were on bosentan monotherapy and 6 patients were on combination therapy: 4 received prostanoids, 1 received sildenafil, and 1 received a prostanoid and sildenafil in combination with bosentan. Seven of the 86 patients were lost to follow-up.
| Treatment at bosentan initiation (number of patients) | All Patients ⁎ (n = 86) | Patients With IPAH/HPAH (n = 36) | Patients With PAH-CHD (n = 48) | ||
|---|---|---|---|---|---|
| monotherapy (11) | plus prostanoid (25) | monotherapy (30) | plus prostanoid (18) | ||
| Continued bosentan | 25 (29%) | 5 (45%) | 7 (28%) | 6 (20%) | 5 (28%) |
| Discontinued | 43 (50%) | 3 (27%) | 15 (60%) | 17 (57%) | 8 (44%) |
| Increase in liver enzymes | 5 (6%) | 0 (0%) | 2 (8%) | 2 (7%) | 1 (6%) |
| Other adverse events | 5 (6%) | 0 (0%) | 2 (8%) | 3 (10%) | 0 (0%) |
| Transplantation | 1 (1%) | 0 (0%) | 1 (4%) | 0 (0%) | 0 (0%) |
| Lack of adequate effect | 19 (22%) | 1 (9%) | 6 (24%) | 6 (20%) | 6 (33%) |
| Pulmonary arterial hypertension worsening † | 9 (10%) | 2 (18%) | 3 (12%) | 3 (10%) | 1 (6%) |
| Other reasons | 4 (5%) | 0 (0%) | 1 (4%) | 3 (10%) | 0 (0%) |
| Death while on bosentan ‡ | 11 (13%) | 1 (9%) | 3 (12%) | 4 (13%) | 3 (17%) |
| Loss of follow-up | 7 (8%) | 2 (18%) | 0 (0%) | 3 (10%) | 2 (11%) |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


