(1)
Clinica di Cardiologia e Aritmologia – Ospedali Riuniti di Ancona, Via Conca 71, 60126 Ancona, Italy
Keywords
Long QT SyndromeICDSecondary preventionTorsade de pointes25.1 Case Report
A 33-year-old woman was referred to our cardiology department after an aborted sudden cardiac death.
Few days before cardiologic visit, an accidental diagnosis of long QT interval was done. The cardiologist noticed a QT interval of 560 ms with a cardiac rate of 58 bpm. Therefore, a 24-h ECG Holter recording was booked. On the day of the recording, the patient collapsed while standing at the red traffic light. She had a spontaneous recovery of consciousness in a few seconds with no memory left.
The patient was then transferred to the ER at the nearest hospital. She fully recovered without neurological sequelae, and the ECG showed sinus rhythm with a prolonged QT interval (580 ms, 55 bpm). Routine blood tests including electrolytes were all within the normal ranges.
The 24-h Holter ECG recording revealed during syncope an episode of polymorphic ventricular tachycardia that was very likely to be a torsade de pointes with later spontaneous sinus rhythm restoration, despite the presence of many artifacts (Fig. 25.1).
The patient was then transferred to our arrhythmology and cardiology clinic for further evaluation and treatment.
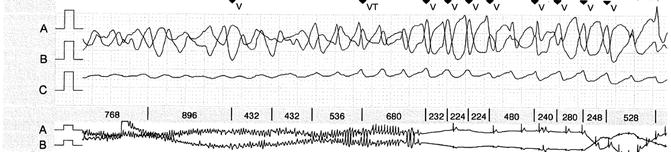
Fig. 25.1
Holter ECG recording showing a torsade de pointes. The smaller trace below shows the spontaneous sinus rhythm recovering
Medical History and Cardiovascular Risk Factors
No cardiovascular risks factor.
No family history of sudden cardiac death or other structural heart diseases.
No history of syncope.
2009: the patient underwent cardiologic visit for nocturnal episodes of palpitations with sudden awakening from sleep and sweating. ECG showed a long QT interval (QTc 560 ms).
Allergies
Dust, cat hair, pollen
Medications
None
Vital Signs
Temperature: 36 °C
Heart rate: 55 bpm
Arterial blood pressure: 100/60 mmHg
Respiratory rate: 18 breaths/min
Oxygen saturation: 100 %
Physical Examination
General: no fatigue, no acute distress; alert, awake, and oriented. Well developed and well nourished
Neck: supple, no jugular venous distention, no lymphadenopathy, and no carotid bruit
Cardiovascular: regular rate and rhythm, S1 and S2 normal, no murmurs, rubs or gallops, point of maximal intensity non-displaced and non-sustained, no hepatojugular reflux, and capillary refill less than 2 s
Lungs: no rales, no rhonchi or wheezes, no egophony, no alterations in tactile fremitus, and normal percussion
Abdomen: no pulsatile masses, normal bowel sounds in all four quadrants, no high-pitched or tinkling sounds, resonant to percussion, soft, non-distended/non-tender, no rebound or guarding, no costovertebral angle tenderness, and no hepatosplenomegaly
Extremities: no cyanosis or clubbing and no peripheral edema
Routine Laboratory Tests Performed
Complete blood count: normal
Cholesterol (total, HDL, LDL) and TG: normal
Hepatic function (GOT, GPT, γ–GT, ALP, total bilirubin, direct, and indirect): normal
Thyroid function (TSH, FT3, FT4): normal
Renal function (creatinine, BUN): normal
Serum electrolytes: potassium 4.4 mEq/l, sodium 138 mEq/l, and magnesium 1.8 mg/dl
Instrumental Examination
The recorded ECG (Fig. 25.2a, b) revealed sinus rhythm with 58 bpm heart rate, normal atrioventricular and intraventricular conduction, and normal ventricular repolarization with a corrected QT interval 600 ms long.
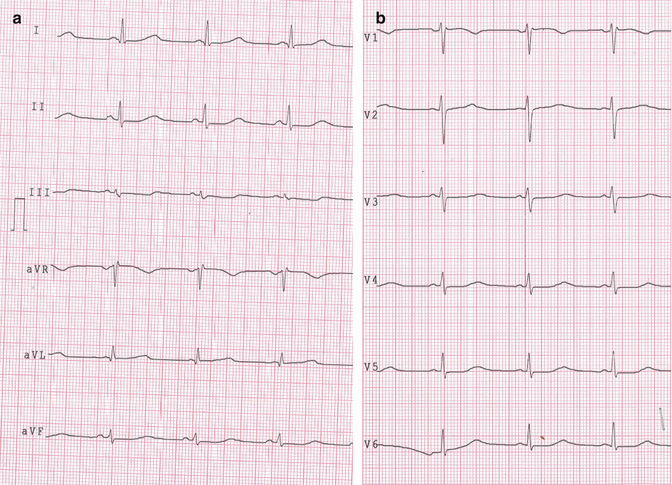
Fig. 25.2
(a, b) Standard rest 12-lead ECG showing a prolonged QT interval (600 ms)
A complete echocardiographic examination was performed and showed normal dimensions and function of cardiac chambers, a mild prolapse of the posterior mitral leaflet with trace of mitral regurgitation, and a mild tricuspid regurgitation with normal pulmonary artery systolic pressure.
No reversible cause of QT interval prolongation was found.
All the family members underwent ECG evaluation, and either the father or brother showed a prolonged QT interval. They both were not taking any medications which could alter the QT interval, and laboratory findings did not show electrolyte abnormalities.
Final Diagnosis
These findings altogether were suggestive for hereditary long QT syndrome. Genetic assessments were therefore performed and it is still underway.
25.2 Long QT Syndrome
Epidemiology
Hereditary long QT syndrome (LQTS) is a congenital disorder characterized by a prolongation of the QT interval detectable on the standard 12-lead ECG with or without T wave [1]. These abnormalities of ventricular repolarization can be present in other family members of the affected subject.
The natural history of LQTS is characterized by the development of life-threatening ventricular arrhythmias such as torsade de pointes (TdP). This arrhythmia can be self-limiting and asymptomatic or they can manifest with syncope or sudden cardiac death (SCD) [2].
The prevalence of this genetic disorder is estimated to be at least 1:5,000 subjects. Considering that the affected subject can be asymptomatic and there are cases of sudden cardiac death with no defined etiology, the real prevalence of LQTS may be considerably higher [1, 2].
Moreover, 10–40 % of affected patients present a nondiagnostic QT interval at rest because of an intermittent nature of the ECG abnormalities or a borderline QT value that make the diagnosis challenging [3].
Long QT syndrome more often manifests before puberty in males and after puberty in females, and the probability of a cardiac event depends on the degree of QT prolongation.
Among untreated symptomatic patients, mortality is high, with 20 % of deaths in the first year after the first cardiac event and approximately 50 % within 10 years [2].
Pathophysiology
Since the discovery of the first LQTS-responsible genes in 1990s, a lot more genes have been related to the syndrome and more than 500 mutations are described nowadays, resulting in 10 different phenotypes (LQTS 1–10).
In about 75 % of patients, mutations involve three main causative genes:
KCNQ1-encoded Kv7.1 channel subunit (IKs potassium channel alpha subunit; LQT1)
KCNH2-encoded Kv11.1 subunit or hERG (IKr potassium channel alpha subunit; LQT2)
SCN5A-encoded Nav1.5 (INa sodium channel alpha subunit, LQT3)
Nearly 70 % of all LQTS are secondary to loss-of-function mutations involving IKs (30–35 %) or IKr (25–40 %) potassium channels, while approximately 5 % result in a gain-of-function mutation involving sodium channel. Less frequently, patients with congenital LQTS present mutation involving:
Ankyrin-B gene: a cytoskeletal membrane adapter which acts as an anchoring protein that binds proteins involved in cardiac electrophysiology and cellular calcium homeostasis (LQT4)
The auxiliary subunits to KCNQ1 (mink; LQT5) or KCNH2 (MiRP1, LQT6)
KCNJ2-encoded Kir2.1 potassium channel with consequent reduction in Kir2.1 current (LQTS7), known as Andersen-Tawil syndrome (ATS), with the phenotype dominated by skeletal abnormalities
CACNA1C-encoded L-type calcium channel subunit with increase in Cav1.2 current and associated with syndactyly in both hands and feet, phenotype known as Timothy syndrome (LQT8)
Cavelolin-3 gene, with increase in late sodium current (LQT9)
SCN4B gene, with increase in late sodium current (LQT10)
In addition, 15–20 % of affected patients present a negative genetic assessment.
The majority of LQTS is inherited as an autosomal dominant trait: the Romano-Ward syndrome affects 1 per 5,000 persons. Sporadic (or de novo) alterations occur 5–10 % of the time. The autosomal recessive form of LQTS, also known as Jervell and Lange-Nielsen syndrome, was firstly described in 1957. It probably affects less than 1 per million people and results from complete loss of Kv7.1 channel function, which precipitates sensorineural deafness in addition to LQT1 or LQT5 mutation [2, 4].
Genotype-Phenotype Correlation
The three main genotypes (LQT1, LQT2, and LQT3) present distinctive clinical phenotypes with important implications in diagnosis, treatment, and long-term clinical course. Indeed, the recognition of gene-specific features can be used to choose the most suitable therapy and to give the correct recommendations to probands and their family. The gene-specific phenotypes include different triggers of ventricular arrhythmias, different ages of symptom onset, and specific ECG morphologies [2, 4].
Patients with LQT1 and less part of LQT2 patients usually present the greater risk of cardiac events after triggers associated with adrenergic stimulation [5]. Life-threatening arrhythmias have been shown to occur under specific circumstances: while diving and swimming for LQT1 patients or acoustic stimuli (such as alarm clock ringing) or sudden awakening for LQT2 patients [4, 6–8].
On the other hand, LQT3 is characterized by the higher risk of events at rest or during sleep.
Patients with LQT3 have fewer events during exercise or stress because they significantly shorten their QTc at high frequencies and therefore they become less susceptible to catecholamine-induced arrhythmias [9]. These findings have consistent clinical implications and explain why these patients have less or no benefit at all from beta-blocker therapy.
Age of first manifestations varies from one genotype to another. In fact, generally, the age of first events is lower in LQT1 than in LQT2 and LQT3 [4].
Some differences can be noticed on the standard 12-lead ECG too. Each one of the three major genotypes (LQT1 to LQT3) seems to have a distinctive T-wave repolarization pattern on the ECG, as shown in Fig. 25.3.
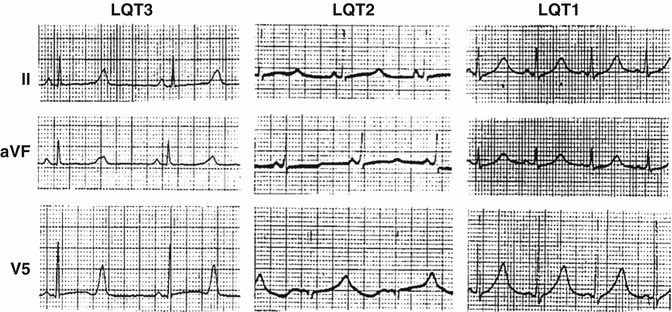
Fig. 25.3
Distinctive ECG patterns of the three main LQTS genotypes. (With permission from Prof. Arthur Moss with the consens of the original publisher (Circulation))
Moreover, every genotype has a different response to exercise. LQT1 patients fail to appropriately shorten their QT interval during exercise and QTc may further lengthen. This effect is due to IKs that in normal subjects is responsible for the abbreviation of the action potential at higher heart rates. On the contrary, patients with LQT3 show shortening of QT interval during exercise. In LQT2, QTc duration behavior is variable [4].
Finally, LQT1 patients have a peculiar response to epinephrine. Epinephrine infusion has an excellent performance in the detection of LQTS1 because it provokes a significant and stable prolongation of the QTc in these patients [10, 12].
Table 25.1 shows the characteristics of different genotypes of LQTS.
Table 25.1
Characteristics of different genotypes of LQTS
Genotype | Higher risk of events | ECG | QT shortening during exercise | Age of first presentation |
|---|---|---|---|---|
LQT1 | Diving/swimming | T waves broad-based with high amplitude | No; may lengthen | Lower |
LQT2 | Alarm clock | Low-amplitude ST-T in the limb leads, biphasic in the precordial leads | Variable | |
LQT3 | Rest/sleeping | Narrow T waves, late-onset peak or biphasic, long isoelectric ST-T segment | Yes |
Clinical Presentation
The long QT syndrome’s main manifestations are electrocardiographic anomalies, syncope, cardiac arrest, and sudden cardiac death.
The ECG anomalies are very different and they depend in part on the type of QT syndrome. The main alteration is the prolongation of the QT interval, but is not always present at rest. In a certain percentage of patients, the QT interval can even be normal (10 % in LQT3 and 37 % in LQT1) [12]. In these cases, the QT interval’s prolongation can be evident during exercise test or infusion of epinephrine [4, 9–11]. Furthermore, repolarization can have strange aspects. For example, notches on the T wave are typical of LQT2.
Syncope, cardiac arrest, and sudden death are the result of ventricular arrhythmias due to the arrhythmogenic condition strictly linked to the genetic and electrical disorders of the disease. These arrhythmias are most of all torsade de pointes VT that become clinically manifest depending on their duration. Sudden death occurs when the arrhythmia is not self-limiting and degenerates into ventricular fibrillation.
Diagnosis
In many cases, the diagnosis of LQTS can easily be done with a standard 12-lead ECG at rest, measuring the QT interval and correcting it with Bazett’s formula (QTc = QT (ms)/√RR(s)). However, there are hidden cases of LQTS in which the QT interval can be normal or borderline at rest (about 20–25 %) [12–16].
Some useful tests have been proposed to unmask the concealed forms of LQTS, and ongoing clinical trials are evaluating and validating them. These tests are:




Exercise test: patients with LQT1 mutation seem to have a more marked QT prolongation during exercise (burst and gradual exercise showed the same results), while those with LQT2 mutation have an exceeding hysteresis of the QT interval (difference between QT interval measured during exercise and 2 min in the recovery phase at similar heart rates) [17–19].< div class='tao-gold-member'>Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
