Figure 12–1.
Predicted topology of Cav1.2 and its resulting proteins related to TS pathophysiology. Mutations found in Timothy syndrome (red dots) interact with channel phosphorylation leading to a sort of hyperphosphorylation state. This process is thought to be mediated by CaMKII (through beta 2 subunit) and by AKAP150, which targets protein kinases to phosphorylation sites (such as Serine 808, 1535 – putative CaMKII site – and 1928)
Beta, alpha2 and delta subunits have a regulatory role. Interestingly, α2 and δ subunits are encoded by a single gene that is translated as a precursor polypeptide and is post-translationally cleaved into the two subunits. The transmembrane δ subunit anchors the α2 protein to the membrane via a single putative transmembrane segment. This association is mediated by disulfide bridges. A range of functional effects has been identified for the associated subunits, including ligand binding, increasing peak currents, modulation of activation and inactivation (increasing the rate of both voltage and Ca2+ dependent inactivation) rates [9, 10].
Calcium Channel Function
Cav1.2 is the most abundantly expressed calcium channel in the ventricular myocytes. It produces a voltage-dependent inward Ca2+ current (ICa) that activates upon depolarization and it is a crucial player in the maintenance of the plateau of cardiac action potential; thus the ICa modulation substantially affects action potential duration. Furthermore, Ca2+ ions play an important role in excitation-contraction coupling, since ICa triggers the release of calcium from the sarcoplasmic reticulum thereby elevating the cytoplasmic Ca2+ to initiate contraction.
Regulation and Tissue Distribution of Cardiac Cav1.2 Channel
The regulation of cardiac Cav1.2 channel activity involves several pathways. Protein kinase A (PKA), Protein kinase C (PKC) and Ca2+-binding protein calmodulin constitute key mechanisms for controlling Ca2+ influx [11–14]. Furthermore, relevant to Timothy syndrome pathophysiology (see below), Cav1.2 channel activity is also sensitive to calcium concentration, catecholamine [15] and Ca2+-Calmodulin-dependent protein kinase II (CaMKII) [16, 17].
Beside cardiac muscle, Cav1.2 is expressed in several tissues including, peripheral and central nervous system, liver, testis, spleen, connective tissue, and bone marrow. It follows that with such a widespread distribution CaV1.2 abnormalities may perturb the function of several organs and cause complex phenotypes.
Another level of complexity of calcium channel physiology is represented by the fact that CACNA1c gene (the gene encoding for Cav1.2) undergoes extensive alternative splicing. More than 20 splice variants have been described, producing expressed peptides with distinct electrophysiological and pharmacological properties [18]. Cell-selective expression of Cav1.2 channels containing specific alternatively spliced exons increases the functional variability for specific cellular activities in response to changing physiological signals. This complex regulation emphasizes the difficulty to predict the clinical manifestation of mutants Cav1.2 proteins. Of clinical relevance, it is unclear why gain of function mutations cause the complex phenotype of Timothy syndrome with multi-organ involvements (see below) while loss of function mutations have been associated with Brugada/short QT phenotype with no signs of extra-cardiac disorders [2].
Timothy Syndrome
Historical Notes
In 1992, Reichenbach reported and defined it as a novel clinical entity, a case of a male infant born at the 36th week of gestation by cesarean section (because of intrauterine bradycardia) who died suddenly at age 5 months. Second degree atrioventricular block, QT interval prolongation and syndactyly were observed [19]. Subsequently Marks et al. reported three additional cases of LQTS and syndactyly [20], confirming the typical features of this disorder.
The first systematic description of the disease was jointly reported in a collaborative study by the groups of Mark Keating and our group. In this study we described 13 cases of this distinctive form of LQTS, presenting with a complex disorder (Fig. 12.2) (see below) with multiorgan involvement and defined the disease as Timothy syndrome (MIM:601005) [5]. Few tens of TS cases have been identified after the initial formal description of the disease, thus suggesting that this condition is extremely rare, possibly because most of the patients die before reproductive age.
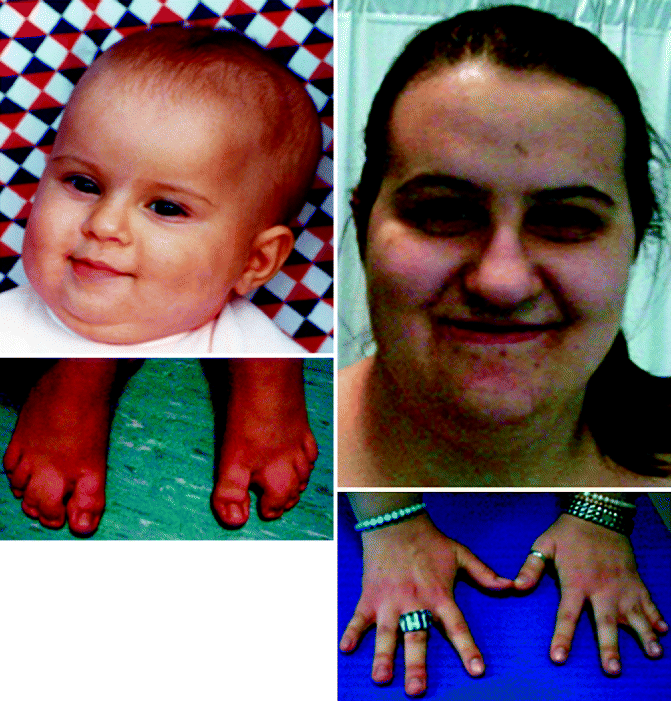
Figure 12–2.
Syndactyly of feet (left panel) and hands (right panel) in patients with TS
Phenotype and Natural History
The first signs of TS may manifest during gestation with fetal bradycardia and 2:1 atrioventricular block but diagnosis is often made within the first few days of life due to abnormal ventricular repolarization and soft tissue syndactyly of hands and toes. With few exceptions (possibly related to a different genetic substrate, see below), syndactyly has been observed in the vast majority of cases.
The QT interval is markedly prolonged in TS (mean value 600 ms); such extreme QT prolongation can cause 2:1 functional AV block. Remarkable abnormalities of T wave morphology consisting of long and straight ST segment, negative T wave and macroscopic T wave alternans, are also evident.
The most frightful manifestations of the disease is represented by cardiac tachyarrhythmia (VT or VF) that occur in 79 % of patients and is the most frequent cause of death (Fig. 12.3). Mortality is in the range of 60 % and the mean age of death is of 2.5 years. However, several additional pathologic (cardiac and extra-cardiac) phenotypes contribute to the TS phenotype:
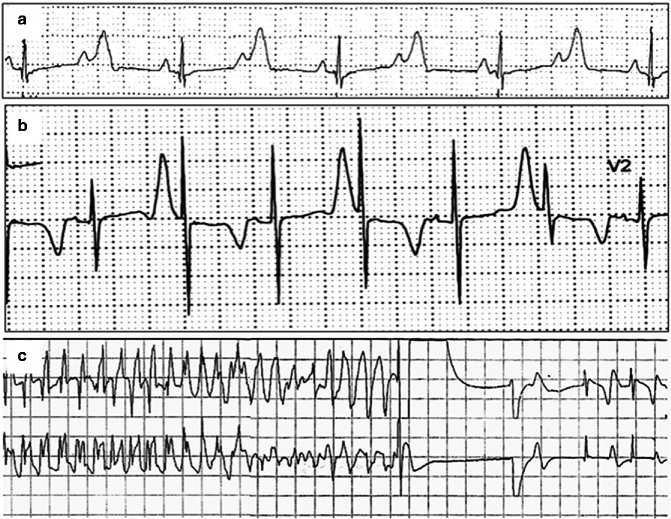
Figure 12–3.
ECG recordings in a TS: (a) long QT and 2:1 atrioventricular block. P = atrial activation, (P wave) (b) T wave alternans. (c) Ventricular fibrillation detected and terminated by ICD shock in a 3 years-old TS patient
congenital heart disease (patent doctus arteriosus, ventricular septal defect, patent foramen ovalis, Tetralogy of Fallot) (55 % of cases).
hypertrophic cardiomyopathy, cardiomegaly and ventricular systolic dysfunction (30 % of cases).
facial dysmorphisms (91 % of cases)
predisposition to sepsis (50 % of cases)
metabolic (severe hypoglycaemia) and immunologic (recurrent infections) disturbances (40 % of cases).
neuropsychiatric involvement (autism and autism spectrum disorder, seizures, psychological developments delays) (83 %).
Genetics of Timothy Syndrome
The ST-T wave morphology in TS patients resembles that of LQTS patients with sodium channel mutations, suggesting the involvement of an inward current active during the plateau phase of the cardiac action potential. However the screening of the SCN5A gene (as well as that of the other known LQTS genes) was negative. Thus CACNA1c was considered a plausible candidate. In 2004, Splawski et al. identified G1216A transition in exon 8A (an alternatively spliced exon), which caused the G406R amino acid transition in DI/S6 in TS patients [5]. Subsequently, Splawski et al. reported two individuals with a severe variant of TS but without syndactyly, they named it as TS2 [21]. Genetic analyses show G1216A and G1204A in exon 8, which caused G406R and G402S amino acid transition respectively (Fig. 12.1).
By means of immunostaining experiments it was shown that exon 8A is expressed in the central nervous system, including hippocampus, cerebellum, and amygdala. Abnormalities of these brain regions have been implicated in autism, a typical feature of TS. Thus, TS may represent the first evidence for a genetic predisposition to behavioral disorders. Interestingly, the clinical relevance of transmembrane Ca2+ current alterations and behavioral abnormalities has been further supported by the association between allelic variants in the CACNA1h gene (CaV3.2, T-type calcium channel) in patients with autism spectrum disorder [22]. Although not directly related with TS pathogenesis these data strengthen the concept that Ca2+ dysfunction may be involved in severe neuropsychological disorders and indirectly the direct the pathogenetic role of Cav1.2 in autism.
Exon 8A of CACNA1c is also expressed throughout the heart and the vascular system, brain, in developing digits and teeth. Thus, the expression pattern of exon 8A is consistent with the phenotypic abnormalities associated with TS [5].
Exons 8 and 8A are mutually exclusive as they encode the same structural domain (DI/S6), but one of the two must be present to encode a functional channel. Experimental data show that 22.8 % of Cav1.2 proteins contain exon 8A, and 77.2 % contain exon 8 [5]. In the brain, 23.2 % contain exon 8A, and 76.8 % contain exon 8. Consistent with the expression, TS2 patients having mutation in exon 8 appear to have a longer QTc than TS1, and a more severe pattern of arrhythmias.
Interestingly the TS2 patient reported so far do not show syndactyly, possibly because of the differential tissue expression of exons 8 and 8A [21]. On the basis of this evidence some work has been done to identify the causes of phenotypic variability in TS. The mechanism of choice between the two exon 8 transcripts is regulated by the polypyrimidine tract binding protein (PTB). This peptide mediates a switch from exon 8 to 8A splicing in a tissue-specific manner and may be responsible for part of variable expressivity [23]. Furthermore somatic mosaicism in the transmission of G406R mutation was demonstrated in the original publication by Splawski et al. [5]. More recently, Etheridge et al. reported that somatic mosaicism contributes to phenotypic variation in TS, suggesting careful genotyping of parental tissue other than peripheral blood lymphocytes [24]. Indeed the possibility for normal parents with no previous family history of TS to generate affected children because of mosaicism has a relevant impact for genetic counseling.
Mechanism of Arrhythmogenesis
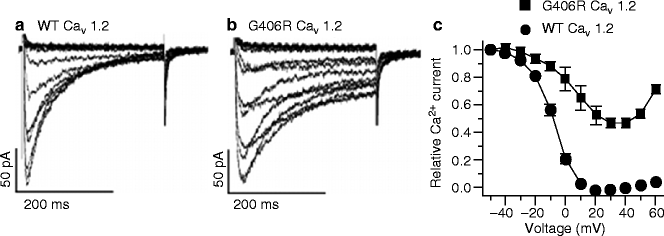
Splawski et al. [5] provided the initial biophysical characterization of TS mutations showing that G406R channels display an abnormal behavior of current inactivation. While inactivation of WT channel current was nearly complete in 300 ms, G406R channels only slightly inactivated during the same time frame. In the voltage dependence of inactivation curves, WT channel inactivation was complete at +20 mV. In contrast, inactivation was only 56 % for the G406R channels (Fig. 12.4). Thus, loss of voltage-dependent inactivation is the main macroscopic current defect leading to action potential prolongation [5, 21]. Such action potential prolongation leads to delayed afterdepolarizations and triggered activity.